The Work page allows you to manage each work that contains Workflow.
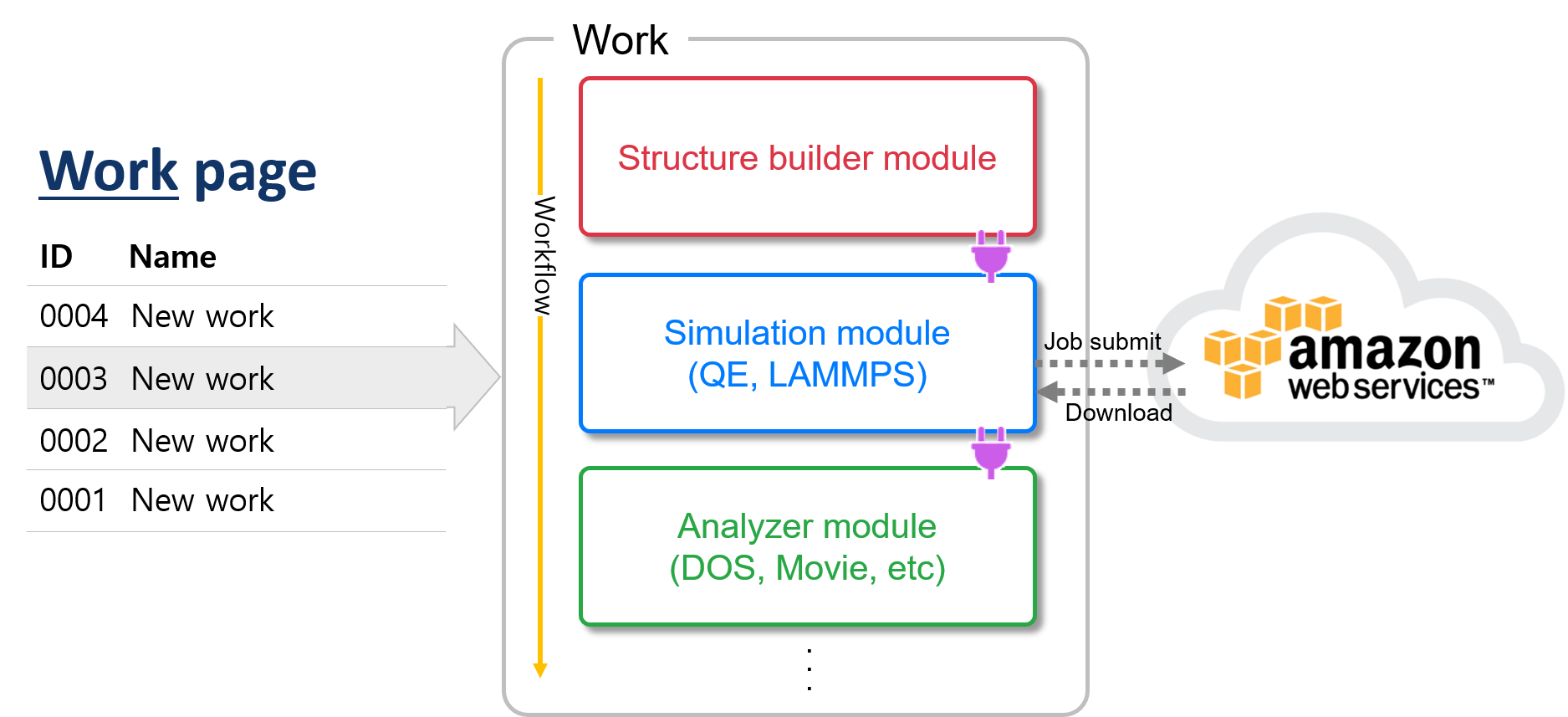
The work can consider as a folder. In other words, one work contains all parts (modules) of a simulation project. It is a different concept to 'Job', the single command submitted to perform the calculation to the MatSQ cloud server, one work can include multiple jobs.
You can simply experience the whole process of material simulation and electronic structure calculation on the work page, from structural modeling to graphical post-processing of results. The currently available software packages are Quantum Espresso, GAMESS for DFT, LAMMPS for MD, Open Calphad for thermodynamic simulation.
Look at the 'Workflow Template' of next section. Add modules according to the workflow template to obtain desired DFT results. Besides, click on the 'New Work' icon to configure the workflow directly. Add modules yourself, and create your own workflow that befits your research.
To use various functions easily, MatSQ modularized many of the functions needed to perform simulation studies. Some modules function independently, but others must be connected to other modules.
The types of modules are as follows:
By default, workflow performs simulations by adding the Simulation module under the Structure Builder and then adding the Analyzer module to view the results graphically. See Modules for more detailed information about each module.

You can see the menu bar, module selector, for adding a Module at the bottom of the Work page. Select to add the desired module. Modules belonging to Simulation and Analyzer must be connected to other modules for use. For information on connections, refer To use modules.
'Connect' is the most important part of configuring workflow in MatSQ. A workflow can be created by assembling multiple modules as if it were assembling blocks. Connect means that bring the information in the selected module becomes available to other modules (The module that clicked the plug icon) as well, and it is a feature that must be used to activating the modules belonging to 'Simulation' and 'Analyzer'. You can connect the two modules through the following method:
icon) as well, and it is a feature that must be used to activating the modules belonging to 'Simulation' and 'Analyzer'. You can connect the two modules through the following method:
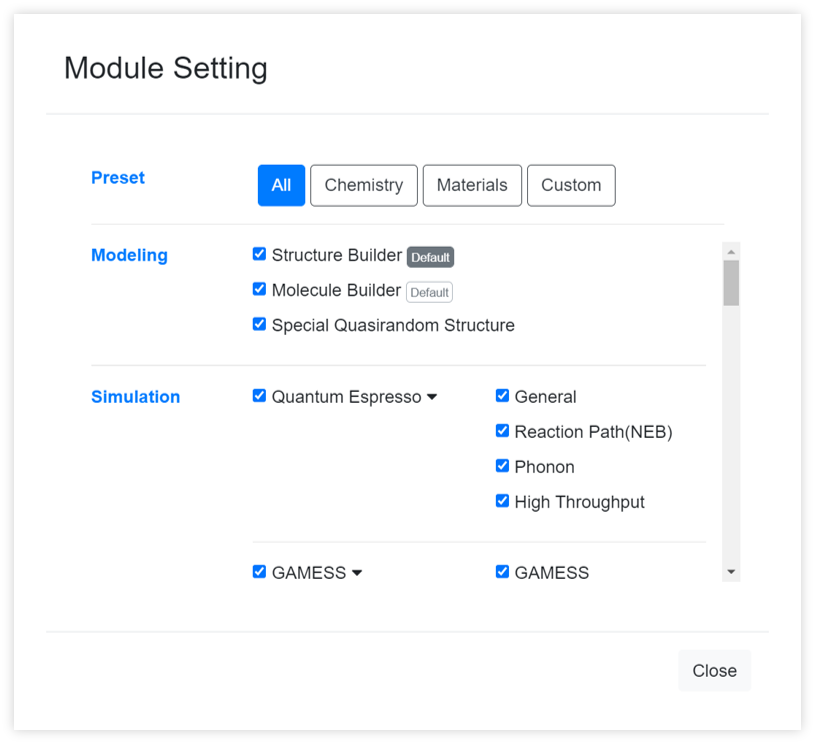

It sets to show only the desired modules among the several modules on the module selector. You can change the default module to be added in first when creating a new work through the button.
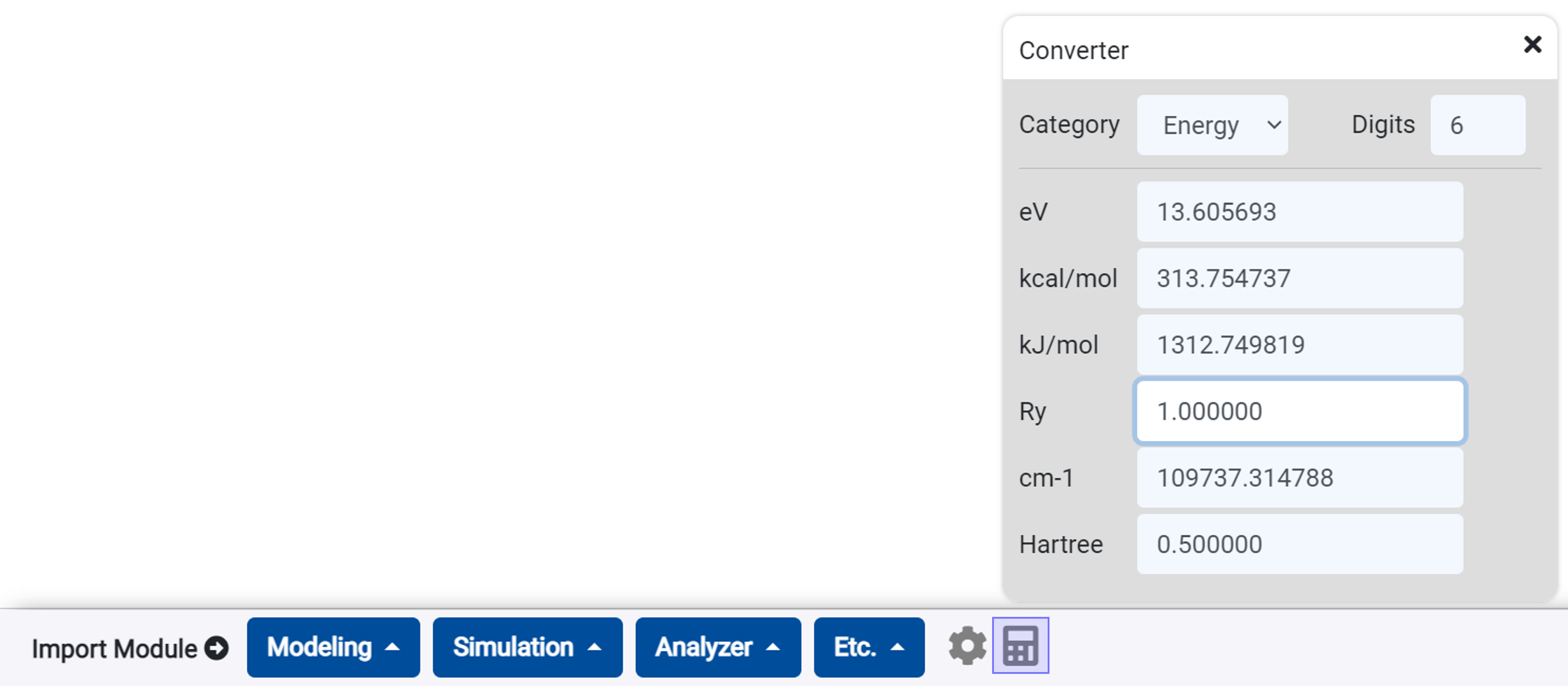
You can convert energy, length units using this converter.
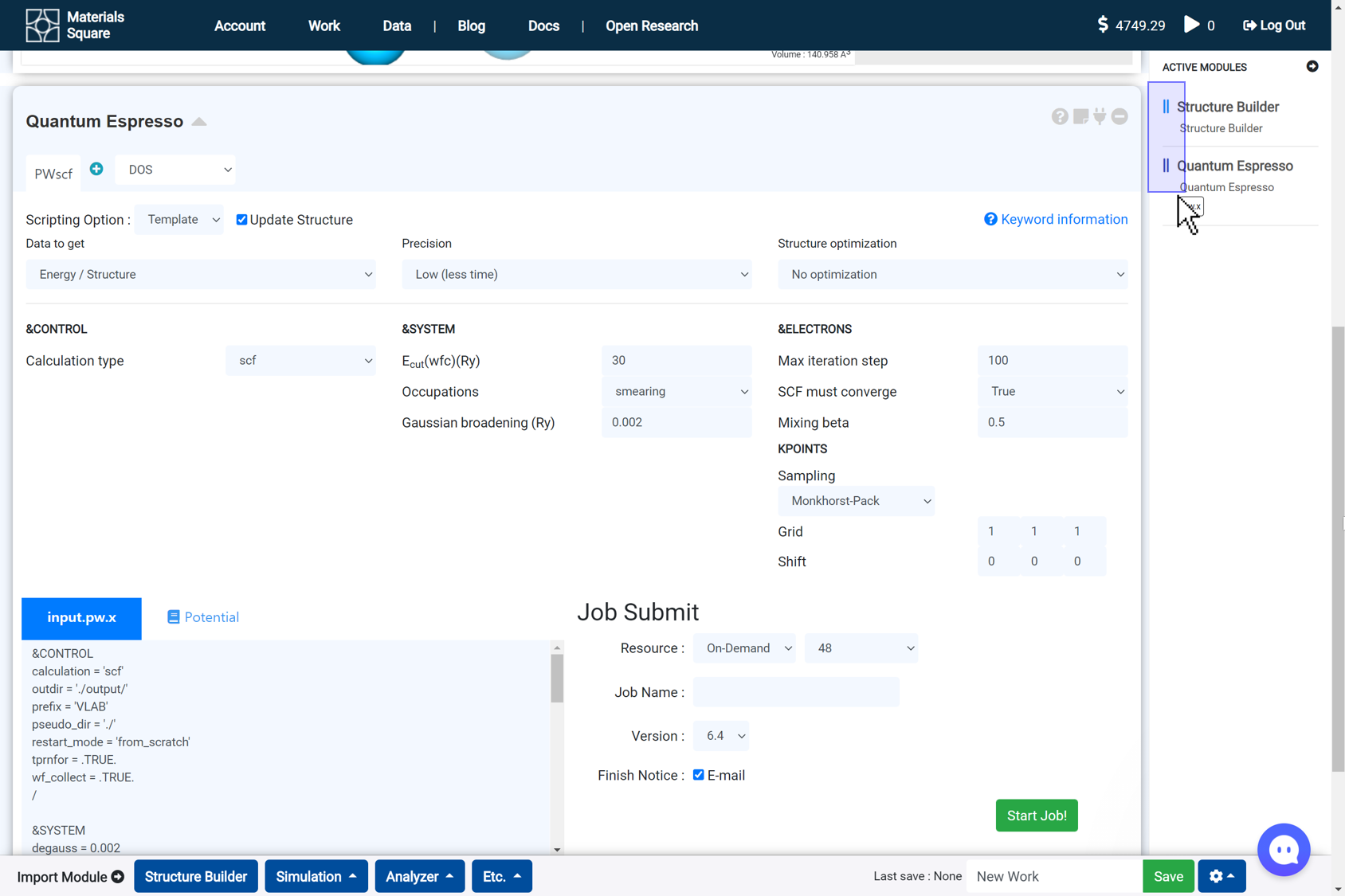
It is ready to start the calculation if you have completed all the settings in the 'Simulation' modules. For the input script setting, please refer to each sections on Modules category.
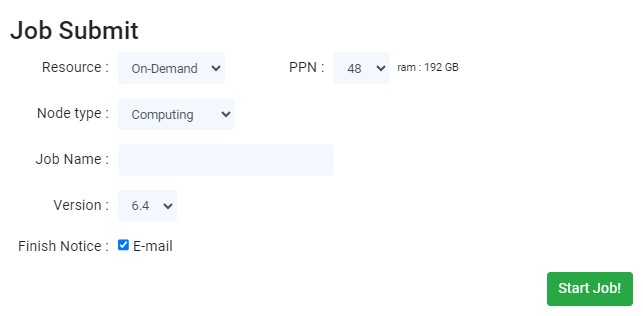
You can select the 'On-demand Instance', and 'Spot Instance' options in the 'Resource' select box. (Spot Instances $0.25/cpu·hour, On-Demand Instances $0.5/cpu·hour)
The 'PPN' refers to the number of cores of parallelization. Up to 48 cores are available in a single node. In this time, the consumed credits are relative to the number of cores.
You can select the 'Computing', and 'High memory' options at the 'Node type' select box. When you are selecting the 'High memory' option, the size of the will be used RAM doubles. Therefore, it is suitable for hybrid functional and phonon calculations. The calculation speed may be slightly reduced.
Enter the job name in the 'Job Name' input. Use a name that is distinguished from other jobs to avoid confusion between one and other calculations.
Select the version of the simulation programs (i.e. Quantum Espresso) at the 'Version'.
Tick in the 'Finish Notice' checkbox if you want to be notified via email after the job termination.
After clicking the button, click the ▶ icon on the top bar to open the dashboard. It shows the progress status of the operation, as shown in the following figure. If you need it, you can pause the job, resume, or stop. The finished/stopped job will be deleted on the list.
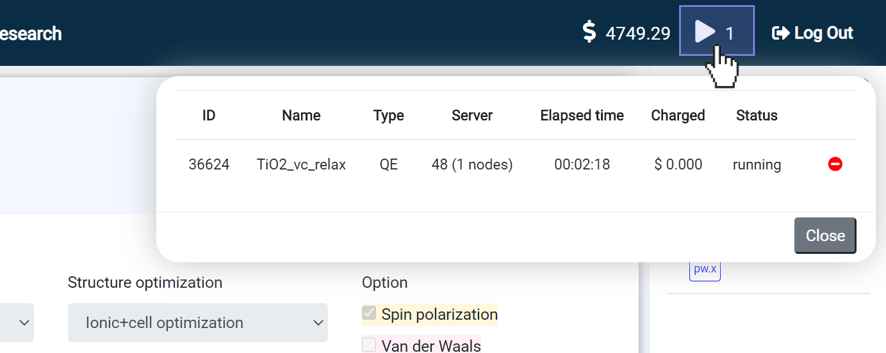
Choosing On-demand Instances results in total CPU hours and credits depleting twice as fast compared to selecting Spot Instances.
Template is a workflow that is prepared for users who are not familiar in calculations.
There are templates available for various workflows. Select an appropriate template for the data you want to obtain.
Follow the steps, and you will be provided with a great guide to getting results quickly and easily.
This template is a workflow prepared to obtain the model’s total energy using DFT. It consists of the following steps:
This template is available to obtain the electronic Density of States (DOS) data of the structure.
For more information, refer to MatSQ's Weekly tip #6 density of states.
This template is designed for easier computation of charge density (electron density) differences. Two charge densities must be calculated in advance to compute charge density differences.
To calculate a band structure, you have to go through several stages of calculations, including the basic DFT calculation (PWscf: scf), the calculation of eigenvalues along high-symmetric points (PWscf: bands), and the calculation to reorder k-points and corresponding eigenvalues (Band Structure). You can calculate the band structure by performing the following steps:
You can simply experience the whole process of material simulation and electronic structure calculation on the work page, from structural modeling to graphical post-processing of results. The currently available software packages are Quantum Espresso, GAMESS for DFT, LAMMPS for MD, Open Calphad for thermodynamic simulation.
Look at the 'Workflow Template' of next section. Add modules according to the workflow template to obtain desired DFT results. Besides, click on the 'New Work' icon to configure the workflow directly. Add modules yourself, and create your own workflow that befits your research.
To use various functions easily, MatSQ modularized many of the functions needed to perform simulation studies. Some modules function independently, but others must be connected to other modules.
The types of modules are as follows:
| The 'Modeling' modules are for creating structure data. They contain the visualizer for identifying, and the manipulators for creating and modifying crystal structures or molecules to perform simulations. | |
| The 'Simulation' modules are for conducting calculations. You can set up the input script and parallel computing options to perform simulations. MatSQ provides a graphical user interface (GUI) for the easy use of Quantum Espresso, GAMESS, LAMMPS, and Open calphad packages. These modules except the Calphad must be connected to a Modeling module or previous finished simulation module to bring necessary data. | |
| The Analyzer module is used to present simulation calculations as graphical charts. It must be connected to the 'Simulation' module to bring the calculation results. | |
| These are modules for convenience. Example is the Memo module to enter additional information. |
By default, workflow performs simulations by adding the Simulation module under the Structure Builder and then adding the Analyzer module to view the results graphically. See Modules for more detailed information about each module.
You can see the menu bar, module selector, for adding a Module at the bottom of the Work page. Select to add the desired module. Modules belonging to Simulation and Analyzer must be connected to other modules for use. For information on connections, refer To use modules.
'Connect' is the most important part of configuring workflow in MatSQ. A workflow can be created by assembling multiple modules as if it were assembling blocks. Connect means that bring the information in the selected module becomes available to other modules (The module that clicked the plug
icon) as well, and it is a feature that must be used to activating the modules belonging to 'Simulation' and 'Analyzer'. You can connect the two modules through the following method:
- Select and add a module from the Module Selector.
- Check that there is a module available for connection.
- 'Connect' can carry out in two methods. First, you can connect with the module if you click a thumbnail image of the module in the right navigation bar. Modules that are not available for connection are translucent.
- If the connection is successful, the color bar appears on the left side of the module name. The module is now ready for use.
▎▎ Currently selected module
▎▎ The modules connected with the selected module
▎▎ The modules indirectly connected with the selected module (The modules connected with the blue bar ▎▎ module)

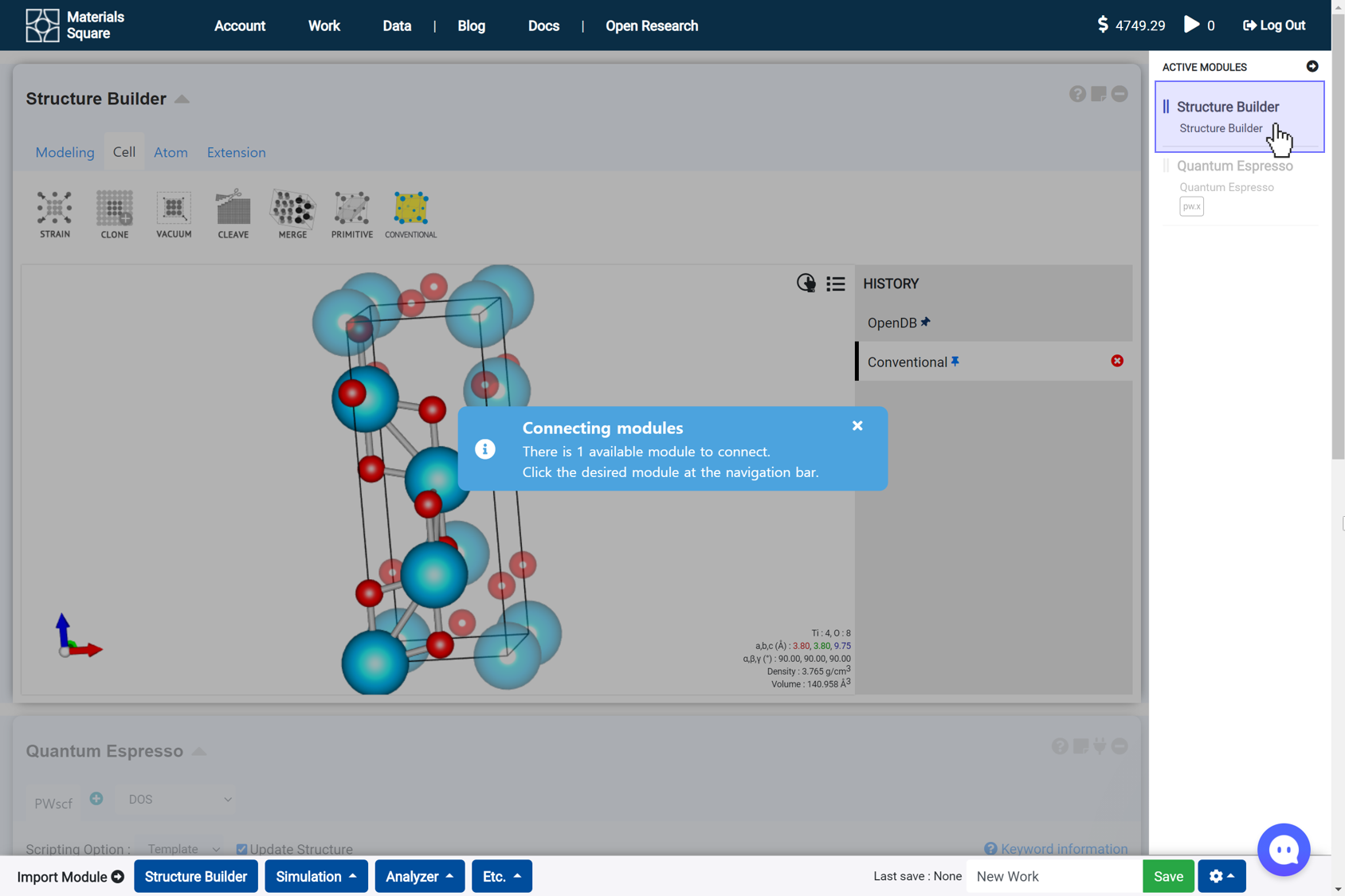
Second, close the connection message and press any part of the module you want to connect.
If you want to exit Connect Mode, press the ESC key.
| Module Name | Click on the module name to rename it. |
| Documentation | Click this button to read a relavent manual. |
| Move | On the right sidebar, drag and drop the thumbnail of a module to move the module. |
| Fold | If the scrollbar is too long, press the ▲ button next to the module name to fold, and reduce the scrollbar. |
| Simple memo | Enter a short memo in the module. |
| Connect module | Press this icon, and click on any part of a module to connect two modules. |
| Delete module | Press this button to delete a module. Deleting the parent module also deletes the linked sub-modules. If you want to cancel this operation, refresh the page to load the last saved state. |
It sets to show only the desired modules among the several modules on the module selector. You can change the default module to be added in first when creating a new work through the button.
You can convert energy, length units using this converter.
It is ready to start the calculation if you have completed all the settings in the 'Simulation' modules. For the input script setting, please refer to each sections on Modules category.
You can select the 'On-demand Instance', and 'Spot Instance' options in the 'Resource' select box. (Spot Instances $0.25/cpu·hour, On-Demand Instances $0.5/cpu·hour)
The 'PPN' refers to the number of cores of parallelization. Up to 48 cores are available in a single node. In this time, the consumed credits are relative to the number of cores.
You can select the 'Computing', and 'High memory' options at the 'Node type' select box. When you are selecting the 'High memory' option, the size of the will be used RAM doubles. Therefore, it is suitable for hybrid functional and phonon calculations. The calculation speed may be slightly reduced.
Enter the job name in the 'Job Name' input. Use a name that is distinguished from other jobs to avoid confusion between one and other calculations.
Select the version of the simulation programs (i.e. Quantum Espresso) at the 'Version'.
Tick in the 'Finish Notice' checkbox if you want to be notified via email after the job termination.
After clicking the button, click the ▶ icon on the top bar to open the dashboard. It shows the progress status of the operation, as shown in the following figure. If you need it, you can pause the job, resume, or stop. The finished/stopped job will be deleted on the list.
Choosing On-demand Instances results in total CPU hours and credits depleting twice as fast compared to selecting Spot Instances.
Template is a workflow that is prepared for users who are not familiar in calculations.
There are templates available for various workflows. Select an appropriate template for the data you want to obtain.
Follow the steps, and you will be provided with a great guide to getting results quickly and easily.
This template is a workflow prepared to obtain the model’s total energy using DFT. It consists of the following steps:
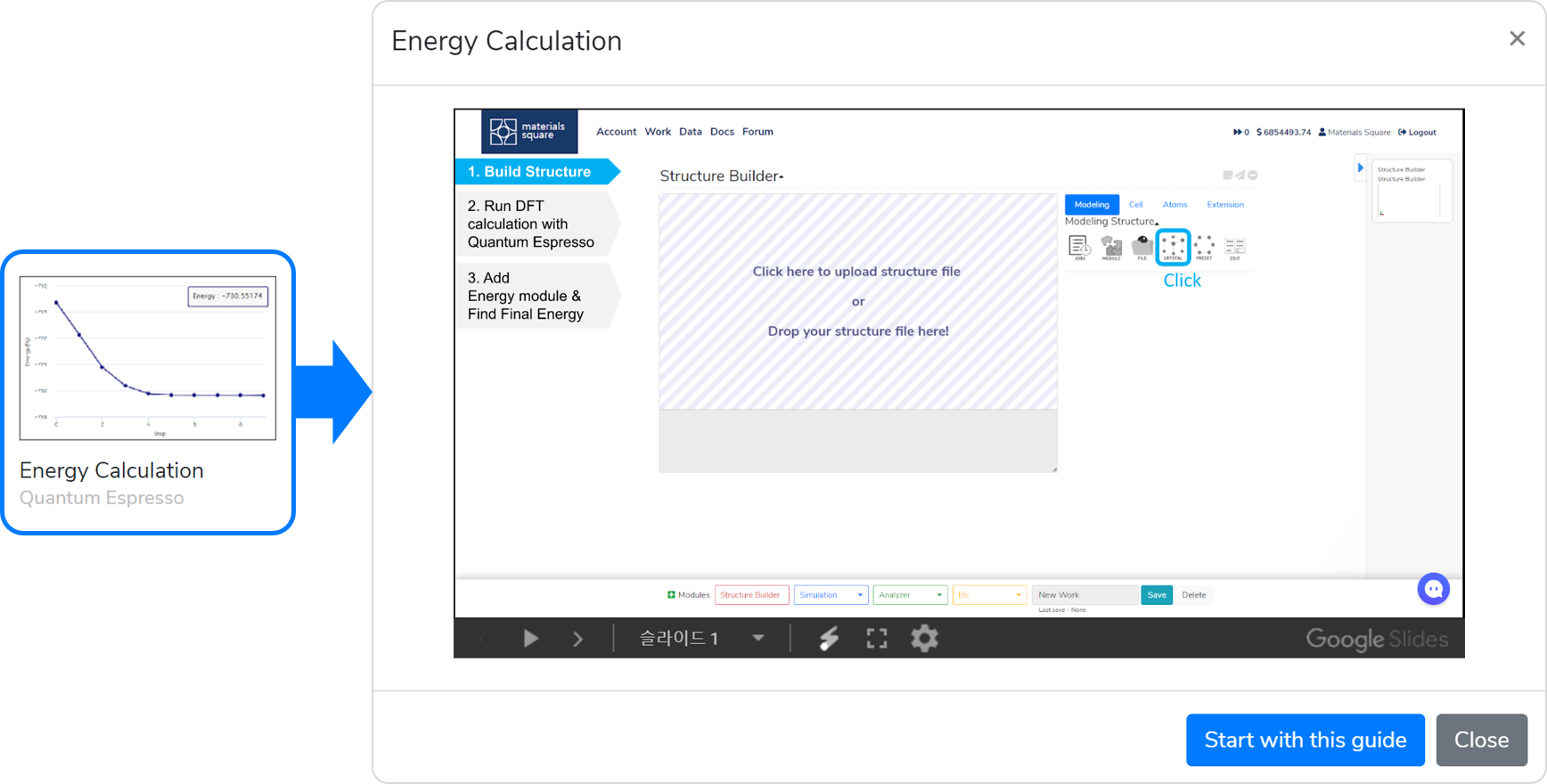
- Model a structure to obtain energy data in the Structure Builder module.
- When you are finished with modeling, add the Quantum Espresso module, and connect it to Structure Builder.
- Answer the questions in the Quantum Espresso module to select input parameters, set the Job name, and click on the button to submit the job.
- After completing the calculation, press the Update button and check that the
message appears.

- Check the energy of the calculated model by adding the Energy module and connecting it to the QE module.
This template is available to obtain the electronic Density of States (DOS) data of the structure.
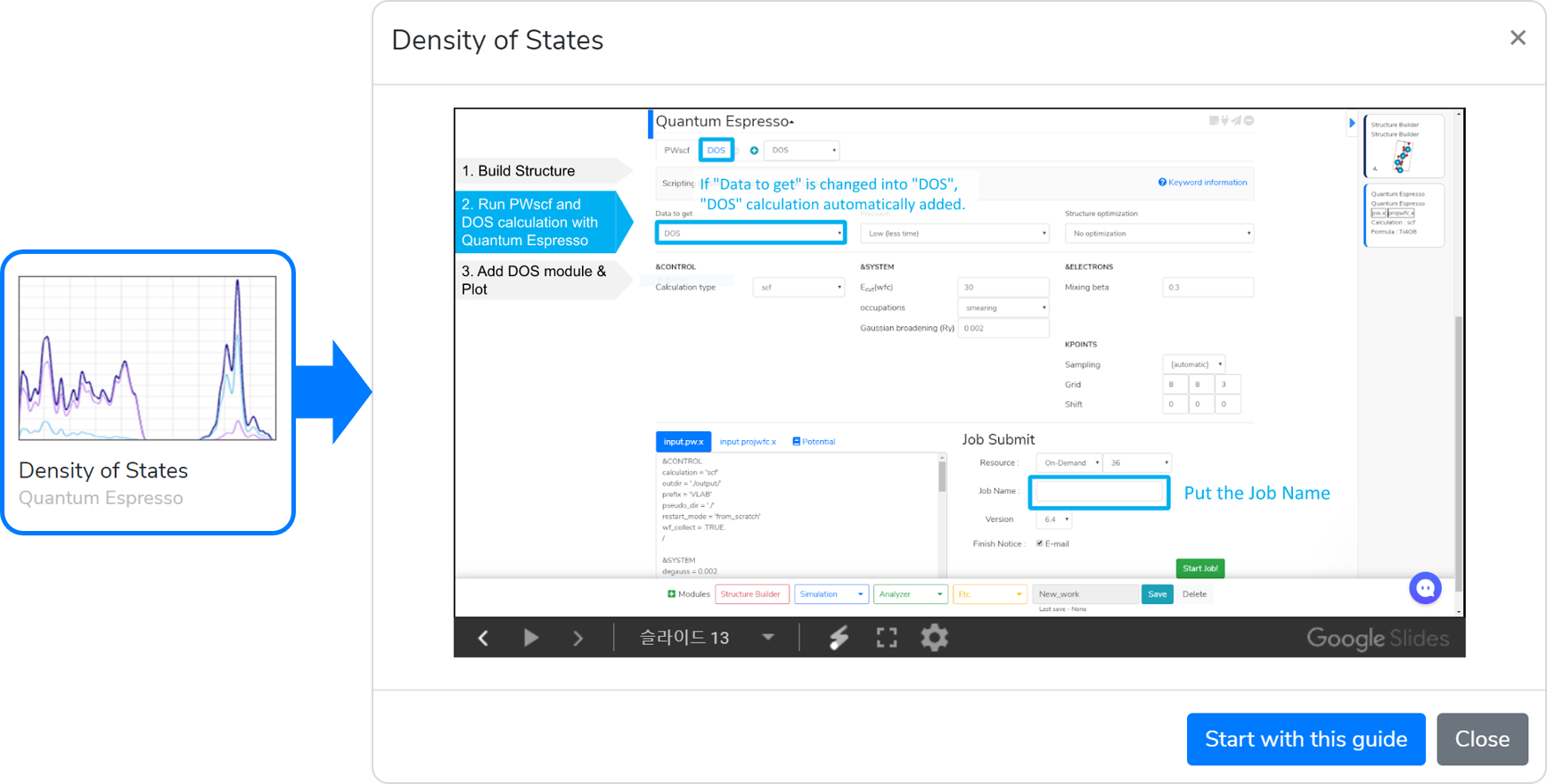
- Model a structure to obtain DOS data in the Structure Builder module.
- When you are finished with modeling, add the Quantum Espresso module, and connect it to Structure Builder described in Step 1.
- Answer the questions in the Quantum Espresso module to select input parameters. Select DOS in “Data to get” question to add the DOS Solver tab.
- Set the Job name, and click on the button to submit the job.
- After completing the calculation, press the Update button and check that the
message appears.
- Add the DOS module, and connect it to the QE module. Then, select atoms and orbitals and press the Add button to check the DOS data.
For more information, refer to MatSQ's Weekly tip #6 density of states.
This template is designed for easier computation of charge density (electron density) differences. Two charge densities must be calculated in advance to compute charge density differences.
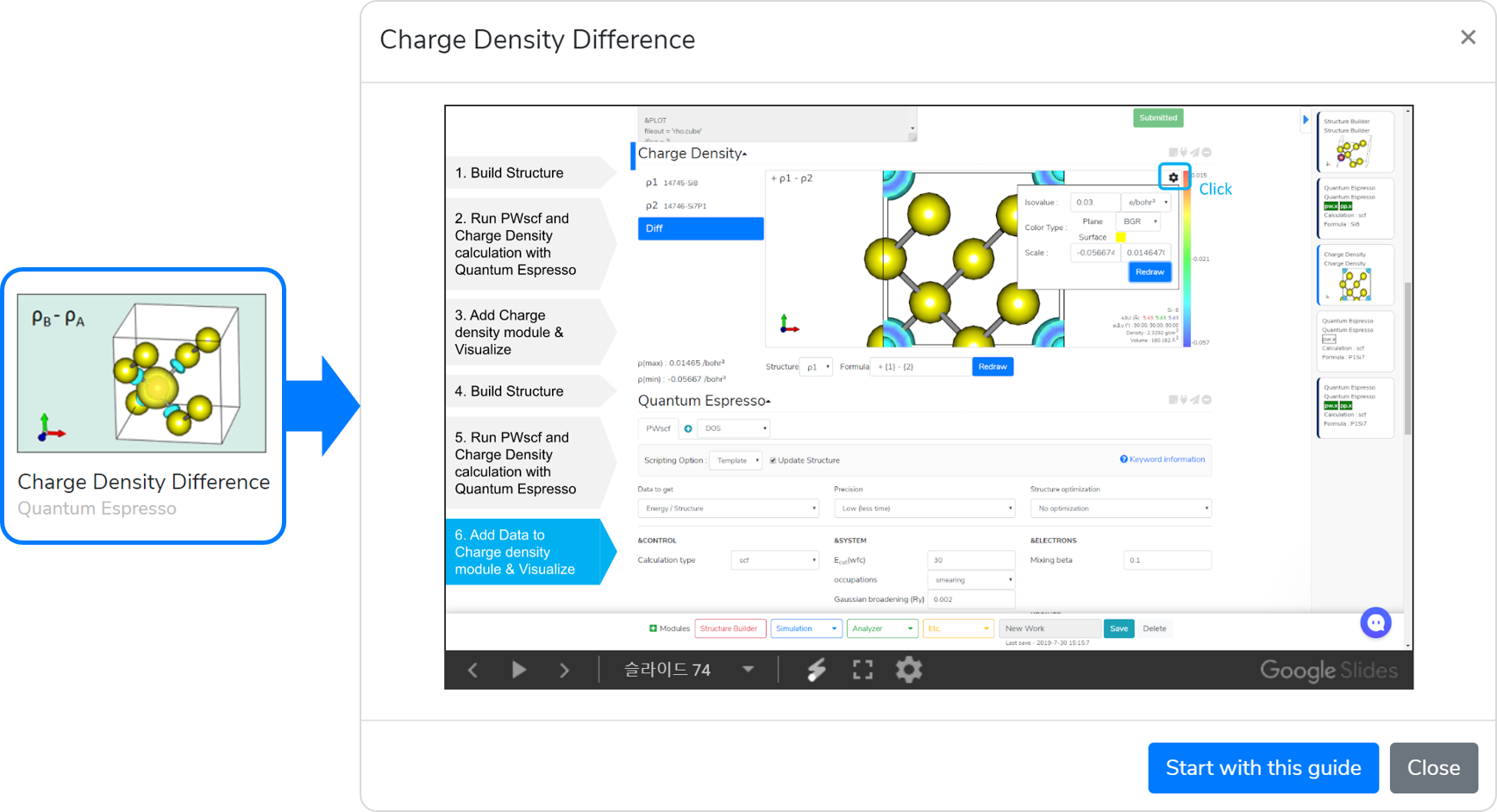
- Create the first structure in the Structure Builder module.
- Answer to the questions in the Quantum Espresso module to select input parameters. In the “Data to get” question, selecting Charge Density adds the Charge Density solver tab. On the Charge Density tab, select the data you want to calculate.
- Set the Job name, and click on the button to submit the job.
- After completing the calculation, press the Update button and check that the
message appears. You can draw the charge density if the calculation is completed normally.
- Add the Charge Density module, and connect it to the QE module to check that the charge density is calculated correctly.
- Create a second structure in the Structure Builder module.
- Answer the questions in the Quantum Espresso module to select input parameters. In the “Data to get” question, select Charge Density, and select the same data you selected under Paragraph 2.
- Set the Job name, and click on the button to submit the job.
- After completing the calculation, press the Update button and check that the
message appears. You can draw the charge density if the calculation is completed normally.
- Connect the Charge Density that was added under Step 5 to the second QE module to draw the second charge density.
- Now, you can draw the charge density differences. Click on ‘Diff’ to calculate ρ A - ρ BYou can change the isovalue, color bar scale, surface color, and cross-section’s color type in the Settings window.
To calculate a band structure, you have to go through several stages of calculations, including the basic DFT calculation (PWscf: scf), the calculation of eigenvalues along high-symmetric points (PWscf: bands), and the calculation to reorder k-points and corresponding eigenvalues (Band Structure). You can calculate the band structure by performing the following steps:
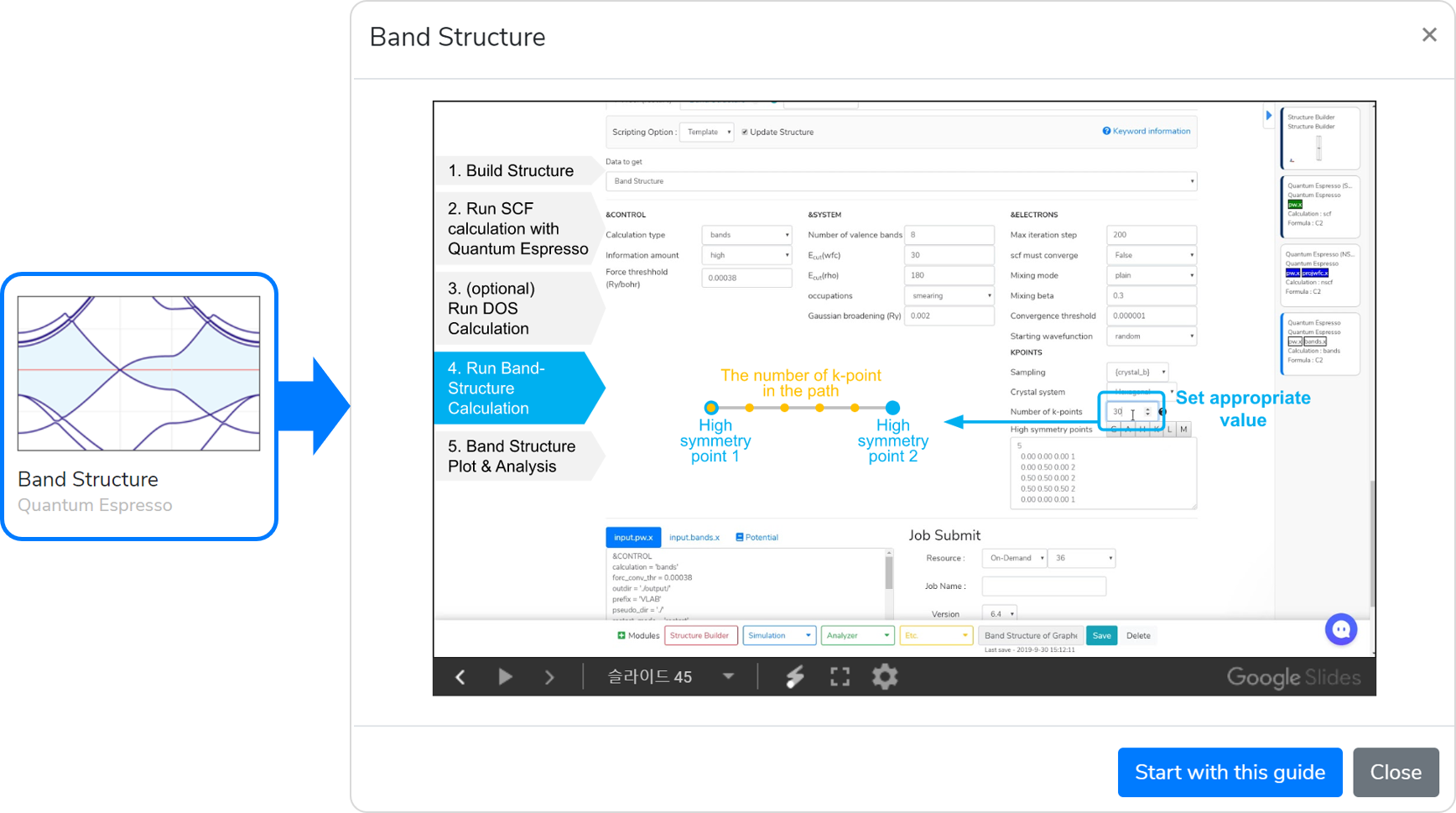
- Model a structure to obtain a band structure in the Structure Builder module.
- Add the Quantum Espresso module in the module selector, Answer the questions in the Quantum Espresso module to select input parameters.
- Set the Job name, and click on the button to submit the job.
- Add a new Quantum Espresso module in the module selector, change the calculation type to “bands,” and set the k-point in QE module.
- Now, the Band Structure solver tab is automatically added. If you have a calculation with spin polarization and want to plot spin up/down separately, you must add two tabs for a spin up/down separately. Press the + button to add a solver for drawing the band structure.
- Set the Job name, and click on the button to submit the job.
- The band structure is now calculated. Add the Band Structure module to draw the band structure.
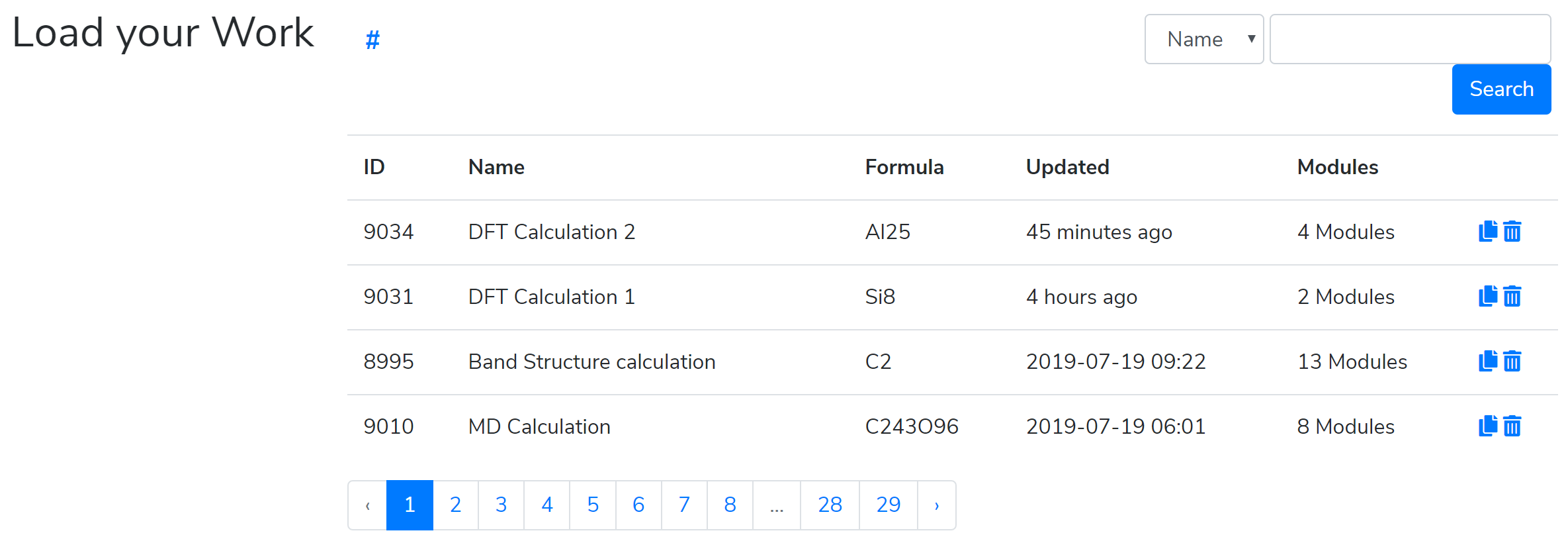
| ID | A unique value for each work that is different from the Job ID |
| Name | The name of a work |
| Formula | A brief view of the structure inside the Structure Builder of the corresponding work page |
| Updated | Shows the last saved time |
| Modules | The type of module inside the corresponding work |
| Clone | Clones the work to create a new one |
| Delete | Deletes the work you chose but not the calculation data |