각각의 기능은 모듈화되어 쉽게 추가하고 관리할 수 있습니다.
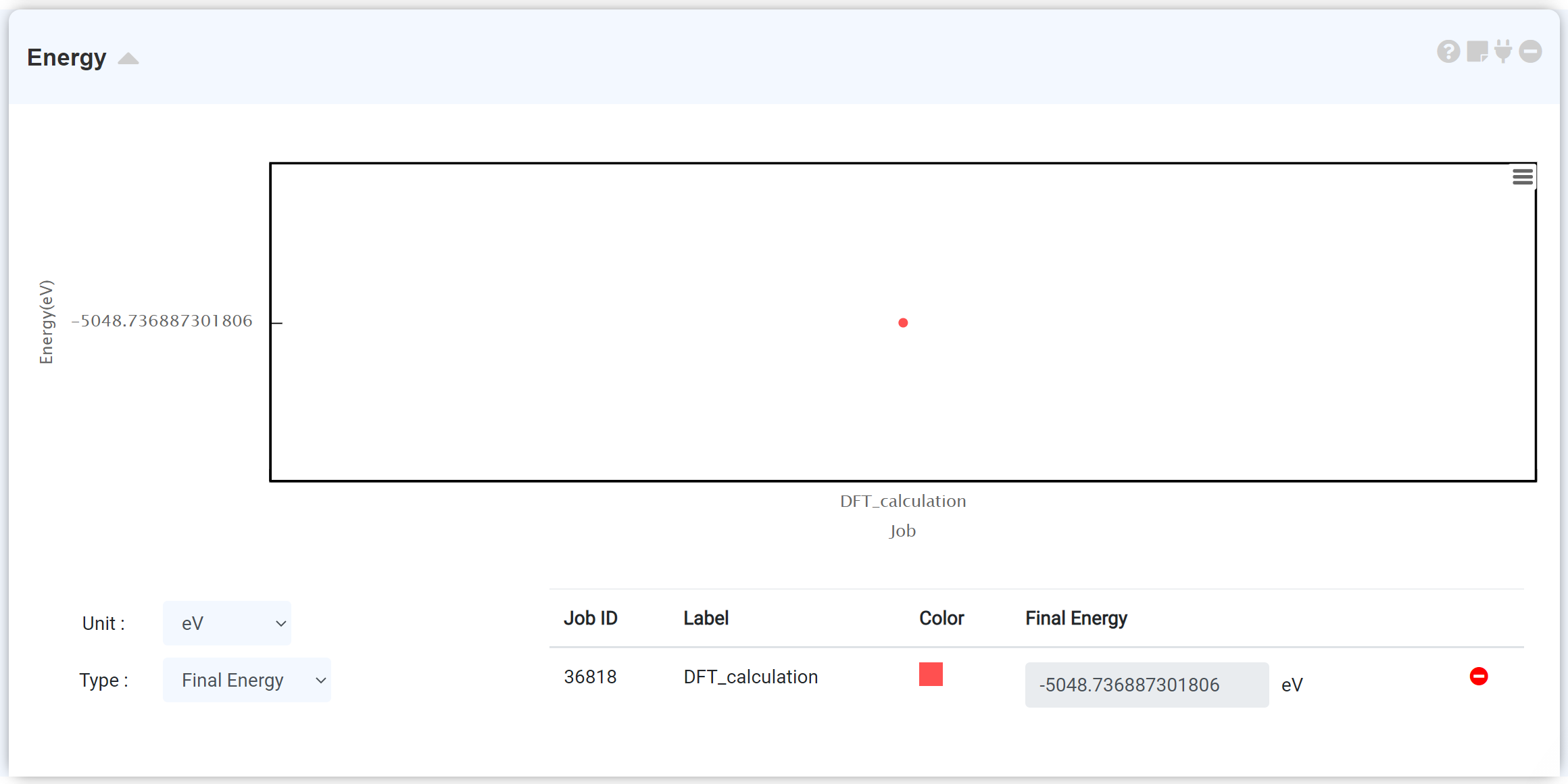
Energy 모듈은 가장 기본적인 분석 도구입니다. 이 모듈을 추가하기 전 시뮬레이션 모듈에서 전자 구조 계산이 선행되어야 합니다. 계산이 종료된 시뮬레이션 모듈에 energy 모듈을 연결하면 에너지 데이터가 표시됩니다.
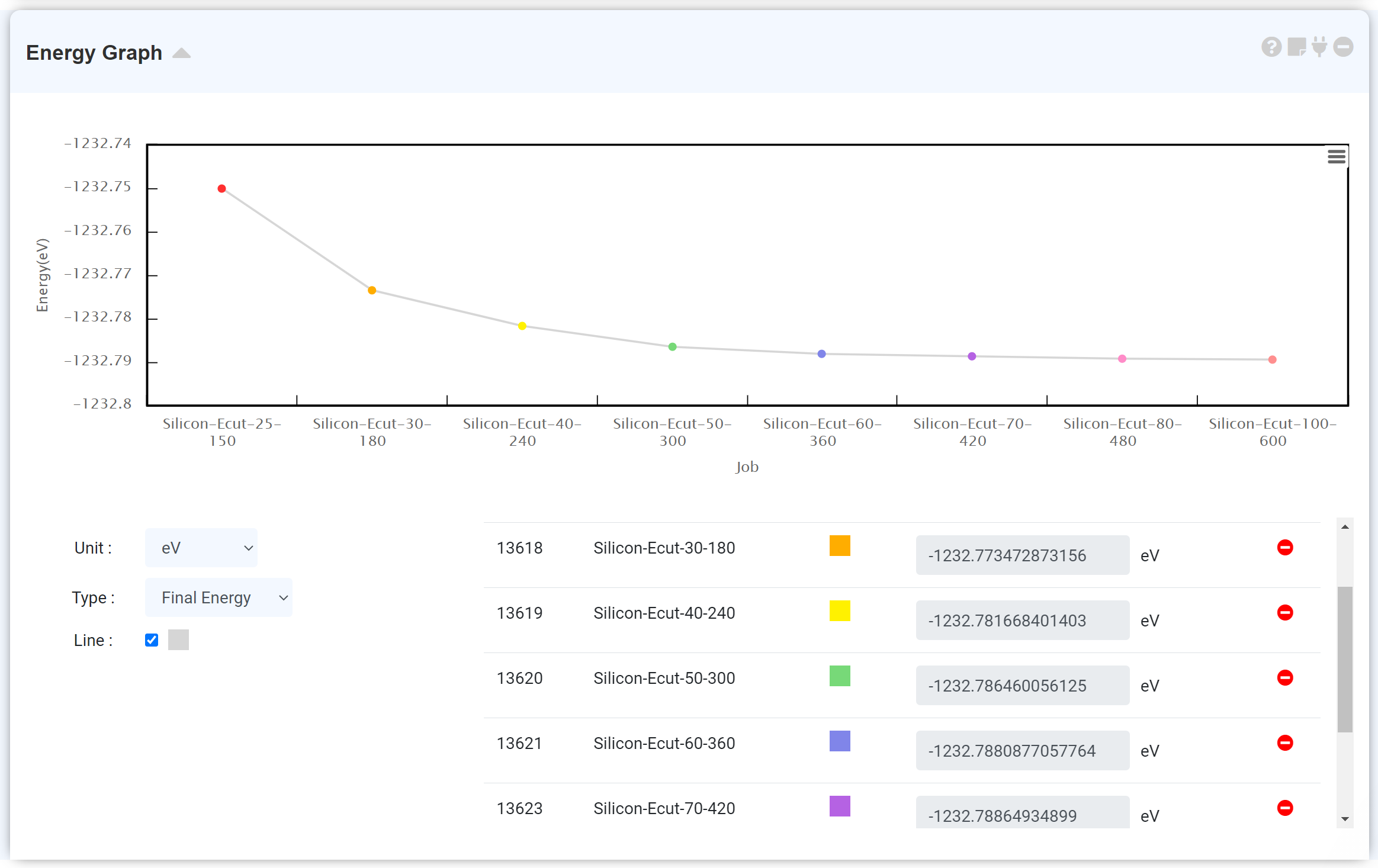
그래프 하단에서 최종 총 에너지를 확인하고, 에너지 단위를 전환할 수 있습니다. y축은 해당 scf 단계에서의 총 에너지이며, x축은 scf 단계를 표시합니다.
하나의 계산 작업은 여러 scf step으로 구성될 수 있으며 하나의 scf step은 iteration step의 집합으로 이루어져 있습니다. Energy 모듈은 iterative self-consistent 계산을 통해 얻어진 총 에너지 값을 도시합니다. 따라서 scf 계산의 경우 단일 scf 계산을 수행하므로 하나의 데이터 지점을 가지며, 여러 scf step으로 이루어진 optimization (QE: (vc-)relax, GAMESS: OPTIMIZE) 계산은 반복되는 scf 계산의 진행에 따라 수렴하는 에너지를 볼 수 있습니다.
Self-consistent calculation에 대해 더 많은 정보는 블로그, Quantum Espresso 또는 GAMESS 공식 홈페이지를 참고하십시오.
에너지 그래프를 확인하는 것은 구조 최적화 과정에서 수렴을 달성했는지 확인하는 좋은 방법입니다. 만일 시뮬레이션 모듈에서 'Successfully finished' 메시지를 확인하였으나 에너지 그래프의 scf 단계가 설정한 최대 scf step 수에 도달했다면, 수렴을 달성하지 못했을 수 있습니다. Restart job (Quantum Espresso) , Restart job (GAMESS) 문서를 참조하십시오.
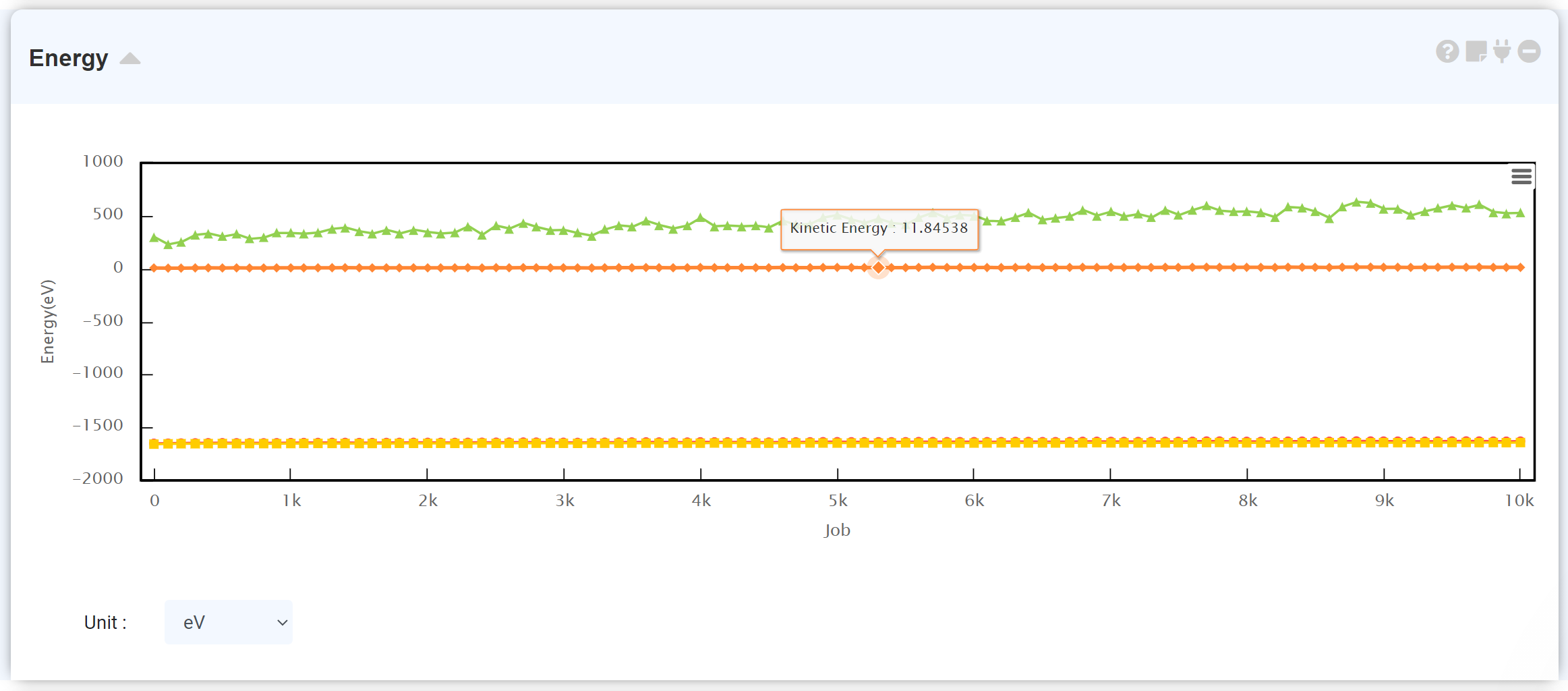
계산이 완료된 LAMMPS 모듈에 에너지 모듈을 연결하면 MD 시뮬레이션 중의 에너지 변화를 확인할 수 있습니다.
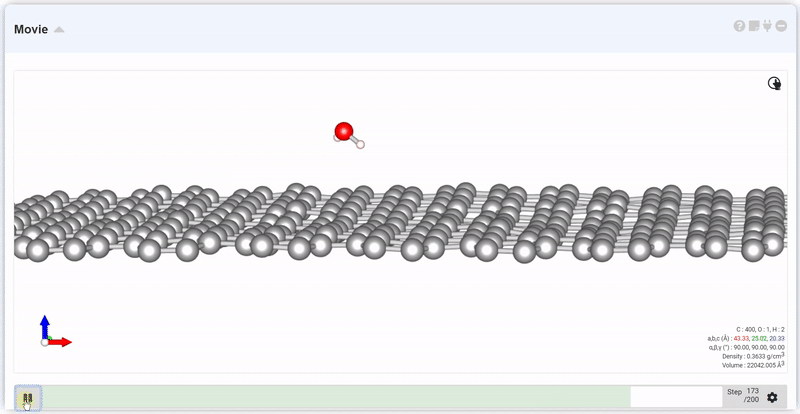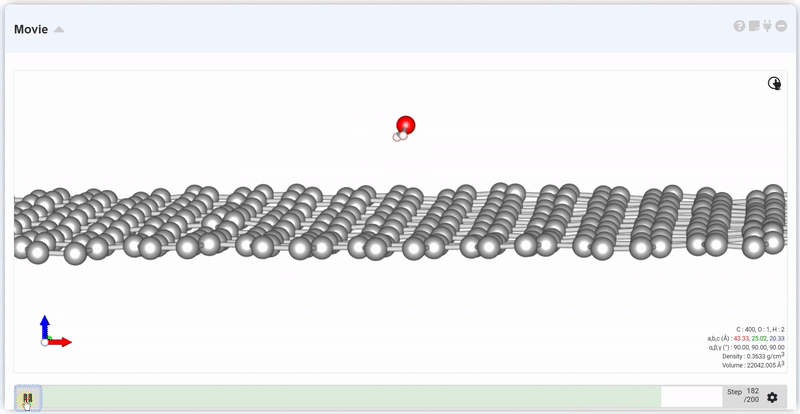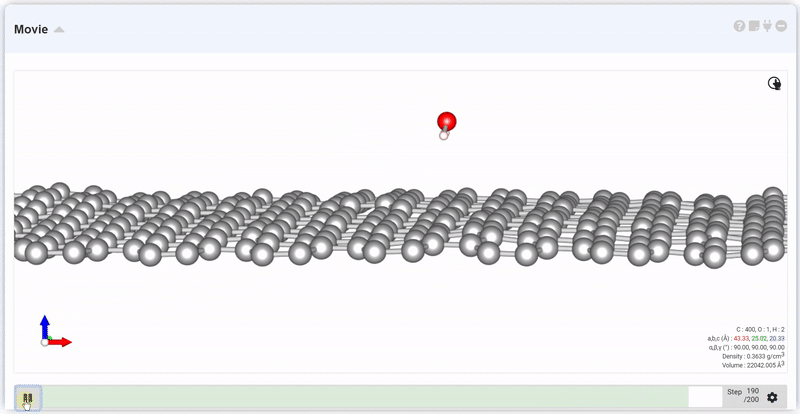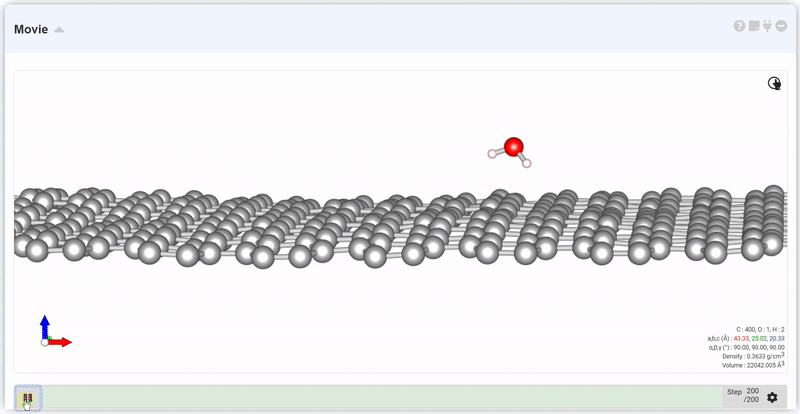
설정창의 Interval에서는 다음 trajectory까지의 시간 간격과 step 간격을 정합니다. Move의 입력창에 원하는 step을 넣고 Move 버튼을 누르면 해당 step에서의 구조를 볼 수 있습니다.
Compare structure 모듈을 사용하면 두 구조를 한 눈에 비교할 수 있습니다. 이 모듈은 구조 정보가 존재하는 모듈 중 Structure Builder, Quantum Espresso, LAMMPS (Custom) 모듈에 연결할 수 있습니다. 모듈 추가 후, 구조를 비교하기 원하는 모듈에 연결하여 선택한 구조를 'Structure A' 에 할당하십시오. 다음으로, 'Structure B' 버튼을 눌러 비교할 구조를 가져오십시오.
Simulation 모듈에 연결하면 '초기 (Initial)' 또는 '최종 (Final)' 구조를 선택할 수 있습니다.
Quantum Espresso 모듈에서 (vc-)relax를 선택하여 계산 모델의 potential energy surface에서 국소 최소값 (local minimum)을 찾는 구조 최적화 (structural optimization / relaxation)를 수행했을 때, 초기 구조와 최종 구조를 비교하여 구조 최적화 과정에서의 구조 변화를 확인할 수 있습니다.
LAMMPS (Custom) 모듈에 연결하면 step 0과 마지막 step에서의 trajectory를 비교할 수 있습니다.
또는 두 개의 서로 다른 구조를 나란히 배치하여 차이를 확인할 수 있습니다.
Compare structure 모듈은 두 개의 visualizer canvas와 데이터 테이블로 구성됩니다. "Sync" 체크박스를 선택하면 두 visualizer의 시점이 동기화됩니다. 표 위에 커서를 올리면 해당 원자가 살구색으로 표시됩니다. 표에서 원자의 이동은 직교 좌표 (Å)로 표시됩니다.
여러 데이터를 입력하고 추세선을 작성하십시오.
'Custom' 옵션에서는 수식을 자유롭게 입력할 수 있습니다. 이때 다음 주의사항을 참고하십시오.
- '# Coeffs. (Number of Coefficients)' 입력창에 변수 x를 제외한 계수의 개수를 정확히 작성하십시오.
- 'Equation' 입력창에 수식을 작성할 때, 곱셈 기호 *를 생략하지 마십시오.
- 'Equation' 입력창에 수식을 작성할 때, 'y='는 작성하지 마십시오.
- 'Equation' 입력창에 수식을 작성할 때, 계수를 나타내는 기호는 a, b, c, d, e, f만 사용할 수 있습니다.
Quantum Espresso 모듈을 이용한 DFT 계산 후, 기본적인 계산 결과 및 NMR 데이터를 확인하기 위한 모듈입니다.
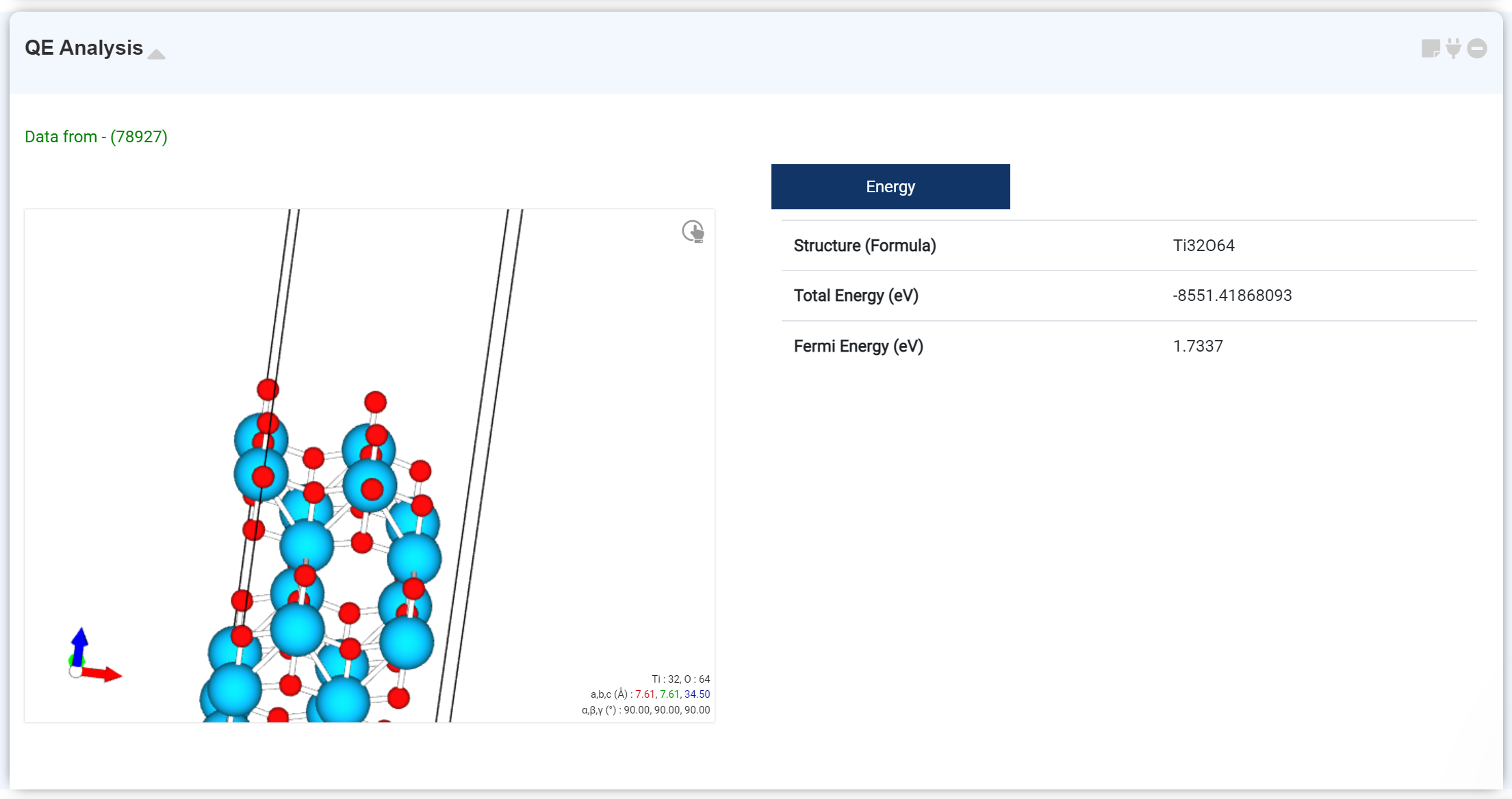
기본적인 계산 결과를 확인할 수 있습니다. 확인할 수 있는 정보는 다음과 같습니다.
- Optimized structure
- Formula
- Total Energy
- Fermi Energy
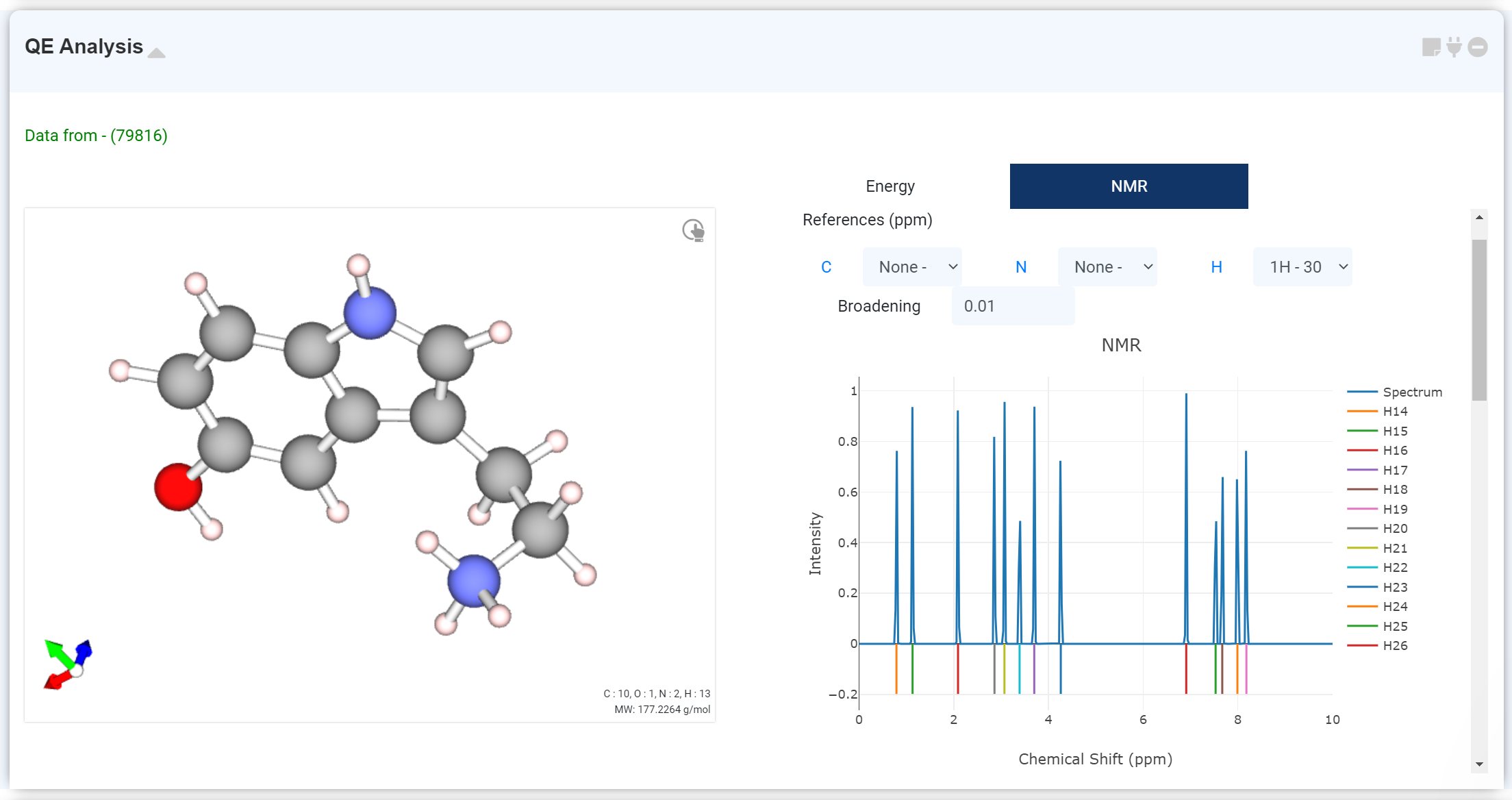
Quantum Espresso 모듈에 NMR solver tab을 추가하여 NMR 계산을 진행하였을 경우, NMR 계산 결과를 확인할 수 있습니다.
스크롤을 내리면 원자 및 계산된 NMR 데이터가 적힌 표를 확인할 수 있습니다. 표의 체크박스에서 원하는 원자를 선택하여 해당 값을 스펙트럼에 추가할 수 있습니다. 또는, 옆의 파란색 원소 기호 링크를 클릭하여 해당 원소의 데이터를 한 번에 확인할 수 있습니다. Select box에서는 x 축 (chemical shift) 값을 보정할 reference를 지정할 수 있습니다. Broadening 값을 수정하여 Spectrum 모양을 조정하십시오.
- Charge Density Difference

- STM Image
Visualizer 우측 상단의 톱니바퀴 버튼을 통해 설정창을 열면 charge density 관련 설정을 바꿀 수 있습니다. 좌측 하단에 위치한 ρ(max), ρ(min) 값을 고려하여 isovalue 값을 바꾸거나, 단면과 표면의 색 그리고 표면색 Gradation scale 값을 바꿀 수 있습니다. Max-Z (Å) 값을 수정하여 Charge Density의 단면을 확인할 수도 있습니다.
2개 이상의 charge density가 연결되었다면 좌측 목록에 각 charge density와 함께 Diff 탭이 활성화됩니다. Diff를 통해 연결된 charge density 값들의 합 혹은 차를 볼 수 있습니다. 자세한 내용은 아래 'Charge Density Difference' 문서를 참고하십시오.
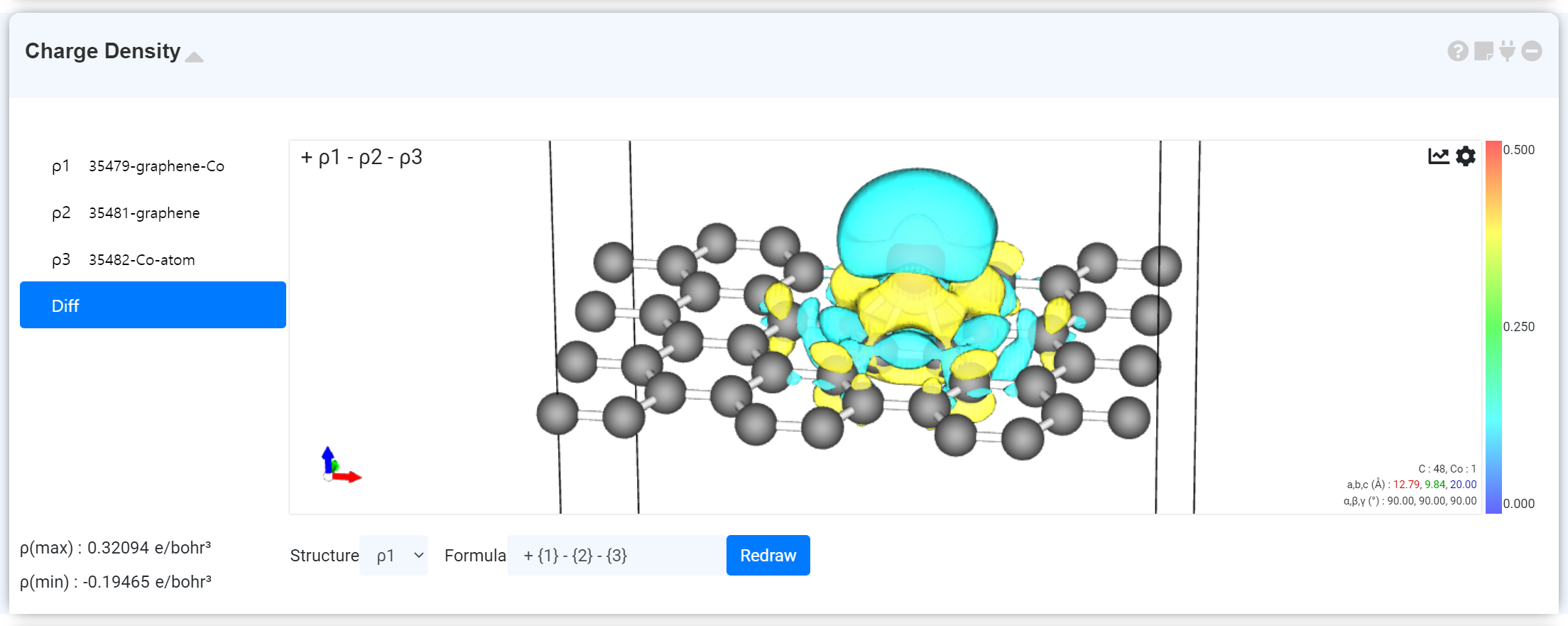
2 개 이상의 Charge density 데이터를 연결하면 Charge density differnce를 확인할 수 있습니다. 좌측의 Charge Density list에서 Diff를 클릭하면 기본적으로 ρ1 구조와 ρ1-ρ2로 계산된 Charge Density 데이터가 표시됩니다. 모듈 하단에 있는 Structure 선택을 변경하여 Visualizer에서 보일 구조를 바꿀 수 있고, Formula를 이용하여 각 값들의 차이를 구할 공식을 정할 수 있습니다. 이때 설정되어 있는 표면의 색과 반대 색을 갖는 표면은 negative 값을 갖는 charge density입니다.
Density of state 그래프는 특정 고유값 (eigenvalue)에 점유된 상태의 수에 대한 정보를 나타냅니다. DOS 그래프는 Energy (eV) 대 states의 개수 (arbitrary units)로 나타내어집니다.
드래그로 확대하여 그래프를 자세히 확인할 수 있습니다. 또한, 우클릭 설정창에서 축 범위와 범례를 설정할 수 있습니다.
DOS 모듈의 visualizer에서 특정 원자만 선택하고 오른쪽의 orbital list에서 특정 오비탈을 선택하면, 해당 원자의 해당 오비탈에 대한 PDOS (projected density of states)를 볼 수 있습니다.
예를 들어, 어떤 원자 혹은 오비탈에 의해 표면 상태 (surface state)가 나타나는지 확인하고 싶을 때, PDOS를 추가하십시오. 'Visualizer canvas' 에서 원하는 원자를 선택하면, 우측 'Orbital list' 에 해당 원자에 해당하는 오비탈만이 표시됩니다. 원하는 오비탈을 선택하여 'Add' 버튼을 누르면 'PDOS' 가 추가됩니다. 이제 어떠한 상태가 어느 화학종에서 기인하는지 확인할 수 있습니다.
DOS는 Band structure diagram과 결합할 수 있습니다 (Band Structure 문서를 참고하십시오). DOS graph의 x축은 band diagram의 y축과 같습니다. 두 그래프의 띠간격은 동일합니다.
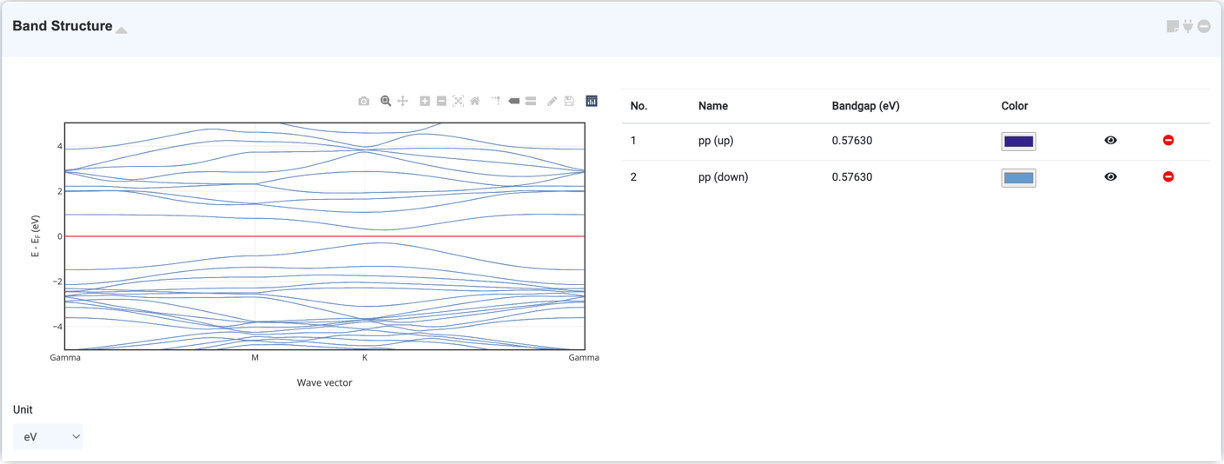
Band structure 모듈은 Quantum Espresso 모듈에서 계산한 Band Structure를 시각화하기 위한 모듈입니다. Band structure를 계산하는 방법은 Band Structure Calculation 문서를 참고하십시오.
Band Structure 모듈을 계산을 완료한 Quantum Espresso 모듈에 연결하십시오.
연결할 수 있는 Quantum Espresso 모듈은 Band structure (bands.x) 혹은 DOS (projwfc.x) 계산이 있는 모듈입니다. 연결 시 Band structure만 있는 계산은 Band structure 데이터로, DOS만 있는 계산은 DOS 데이터로 활용됩니다. 두 가지 계산이 모두 있는 경우는 Band structure, DOS 및 Projected band structure (fatband) 데이터로 지정하여 활용할 수 있습니다.
Band diagram의 x축인 k-path label은 high-symmteric point 중 가장 유사성이 높은 crystal system point set을 찾아 자동으로 설정됩니다. Select box에서 crystal system과 energy 단위를 바꿀 수 있습니다.
DOS 데이터가 있는 Quantum Espresso 모듈을 연결하여 Band structure와 DOS 그래프를 나란히 볼 수 있습니다. Band diagram의 y축은 DOS graph의 x축과 같습니다. 마우스 드래그를 사용해 zoom in/out을 할 수 있고, 그래프 우측 상단에 있는 Reset 버튼을 통해 원래로 돌아갈 수 있습니다. 그래프 하단에 위치한 테이블을 통해 canvas에 그려진 차트를 지우거나 비활성화 할 수 있습니다.
좌측 상단에 위치한 Visualizer와 우측 상단에 위치한 오비탈 셀렉터를 통하여 특정한 원자 및 오비탈에 대해 band structure 및 DOS를 projection하여 보거나 fatband를 그릴 수 있습니다. 자세한 내용은 다음 Projected Band Structure 문서를 참고하십시오.
Band structure를 특정한 원자 혹은 오비탈에 projection하여 fatband를 그리고 싶다면, Band structure를 계산했던 Quantum Espresso 모듈에 'DOS' solver를 추가해서 projection 계산을 수행하십시오.
추가된 데이터는 원 모양 혹은 굵은 선 형태로 표시됩니다. 원의 반경/선의 굵기는 그 점에서 선택된 궤도의 기여도 (contribution)의 합으로 결정됩니다. 기여도는 해당 지점에서의 eigenvalue에 대한 각 orbital의 distribution을 말합니다. 그래프의 색은 동일한 각운동량 양자수 (angular momentum quantum number)를 갖는 선택된 오비탈 색의 평균치입니다.
추가된 데이터가 각운동량 양자수가 다른 여러 데이터를 포함하고 있는 경우, 각 오비탈의 기여도가 색상 그라데이션으로 표시됩니다. 각 오비탈의 기여도를 원의 반지름/선의 굵기로 보고 싶다면 각각의 오비탈을 따로 선택하여 추가하십시오.
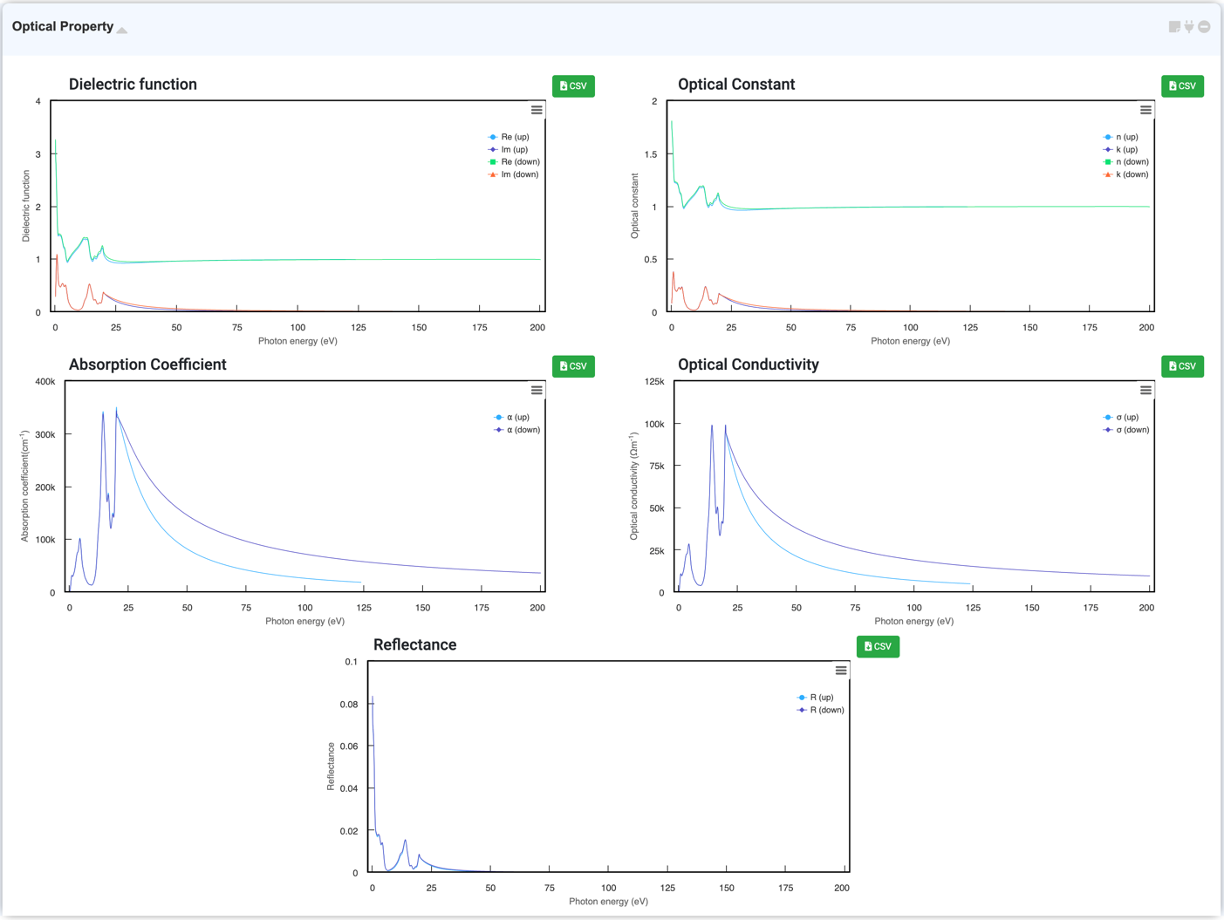
Quantum Espresso 모듈에서 계산한 광학적 특성 데이터를 그래프로 확인할 수 있습니다. Materials Square의 Quantum Espresso 모듈에서 plane-wave basis DFT 계산을 수행하여 여러 광학적 특성 데이터를 얻을 수 있습니다. Dielectric tensor와 joint density of states (JDOS)의 실수부와 허수부를 확인할 수 있습니다. Optical property 계산은 'Norm-Conserving Pseudopotential (LDA and GGA)' 만 지원합니다.
- Dielectric function
- Joint density of state (JDOS)
- Refractive index
- Absorption coefficient
Charge density 모듈은 공간 상에서 동일한 값을 갖는 표면 (isosurface) 및 셀 경계면을 표시하여 계산한 구조의 charge density와 potential을 시각화합니다. Analysis Module에서 Charge Density를 선택 후 활성화된 모듈을 클릭하여 연결하면 charge density 와 potential이 3D/2D 형태로 나타납니다. Visualizer 우측 상단의 톱니바퀴 버튼을 통해 설정창을 열면 charge density 관련 설정을 바꿀 수 있습니다.
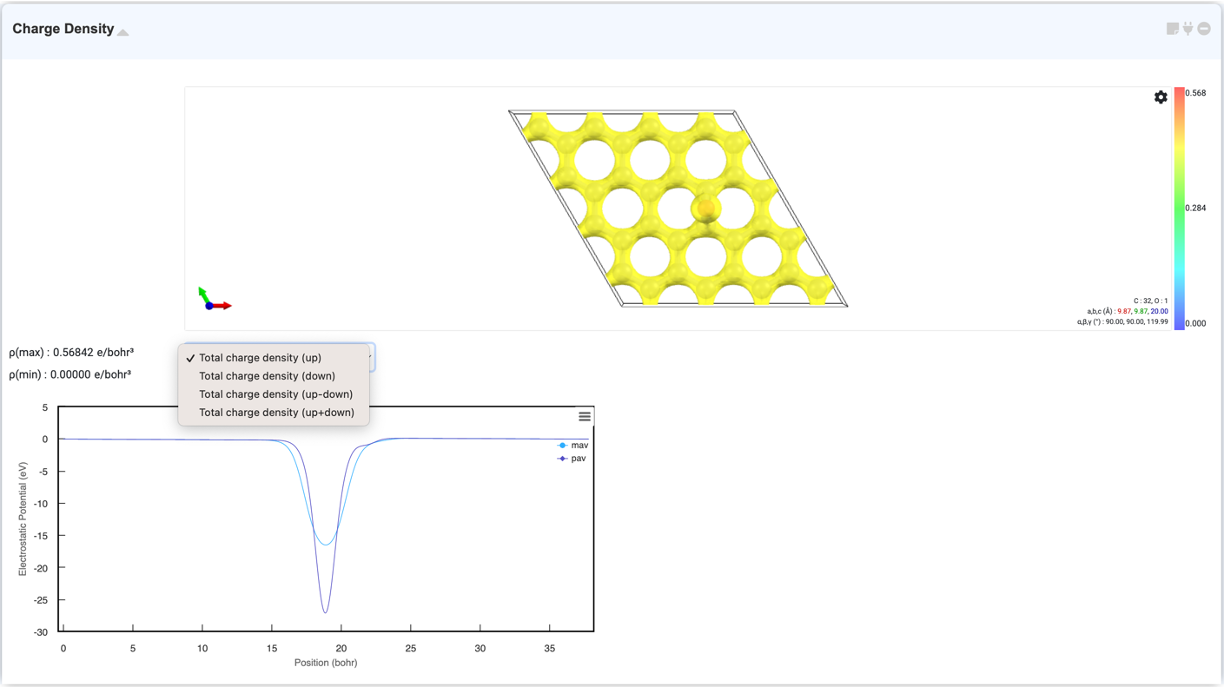
Density of state 그래프는 특정 고유값 (eigenvalue)에 점유된 상태의 수에 대한 정보를 나타내며 그래프는 Energy (eV) 대 states의 개수 (arbitrary units)로 나타내어집니다. Analysis Module에서 “Density of States” 선택 후 활성화된 모듈을 클릭하여 연결하면 DOS 그래프를 확인할 수 있습니다. 드래그로 확대하여 그래프를 자세히 확인할 수 있으며 우클릭 설정창에서 축 범위와 범례를 설정할 수 있습니다.
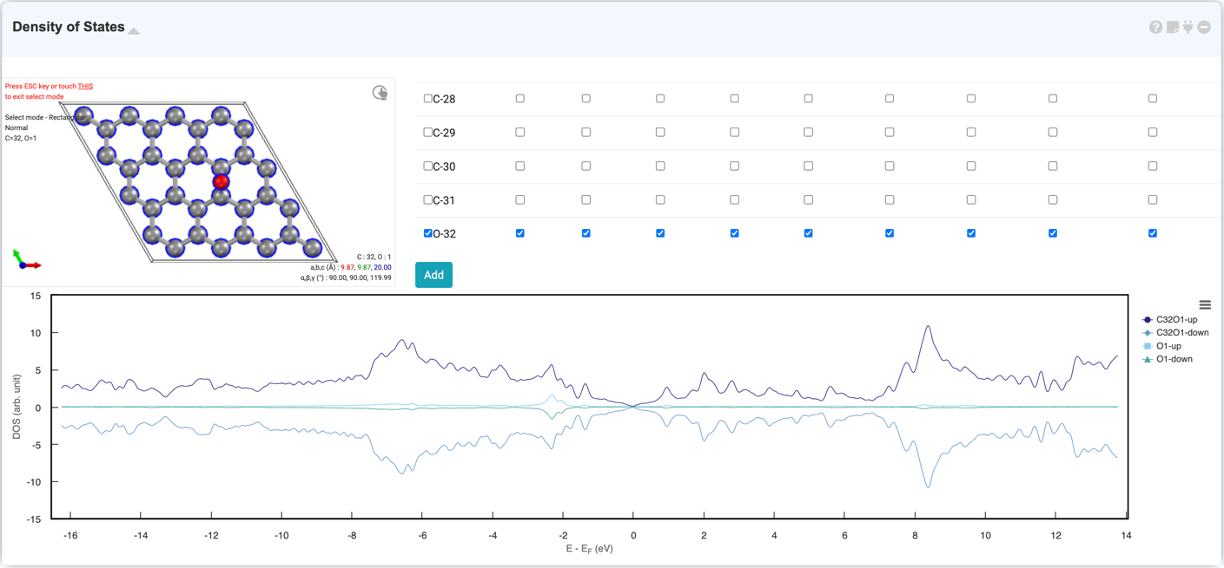
DOS 모듈에서는 특정 원자 오비탈에 대한 PDOS (projected density of states)를 볼 수 있습니다. DOS 모듈 안의 visualizer에서 원하는 원자를 선택하면, 오른쪽의 orbital list에 원자들의 오비탈이 표시가 됩니다. 그 중 원하는 오비탈을 선택하여 'Add' 버튼을 누르면 'PDOS' 가 추가됩니다.
Band structure는 물질의 전자에 허용된 에너지 level, 또는 분포를 나타냅니다. 그래프는 k vector와 energy 로 나타내어집니다. Analysis Module에서 “Band Structure” 선택 후 활성화된 모듈을 클릭하여 연결하면 band structure를 확인할 수 있습니다. 상단의 도구를 통해 확대, 범위 설정, 다운로드 등을 할 수 있습니다.
Analysis Module에서 “Optical Property” 선택 후 활성화된 모듈을 클릭하여 연결하면 시스템의 광학 특성을 그래프로 확인할 수 있습니다. Dielectric function, Optical Constant (refractive index, extinction coefficient), Absorption coefficient, Optical conductivity, Reflectance 를 확인하실 수 있습니다.
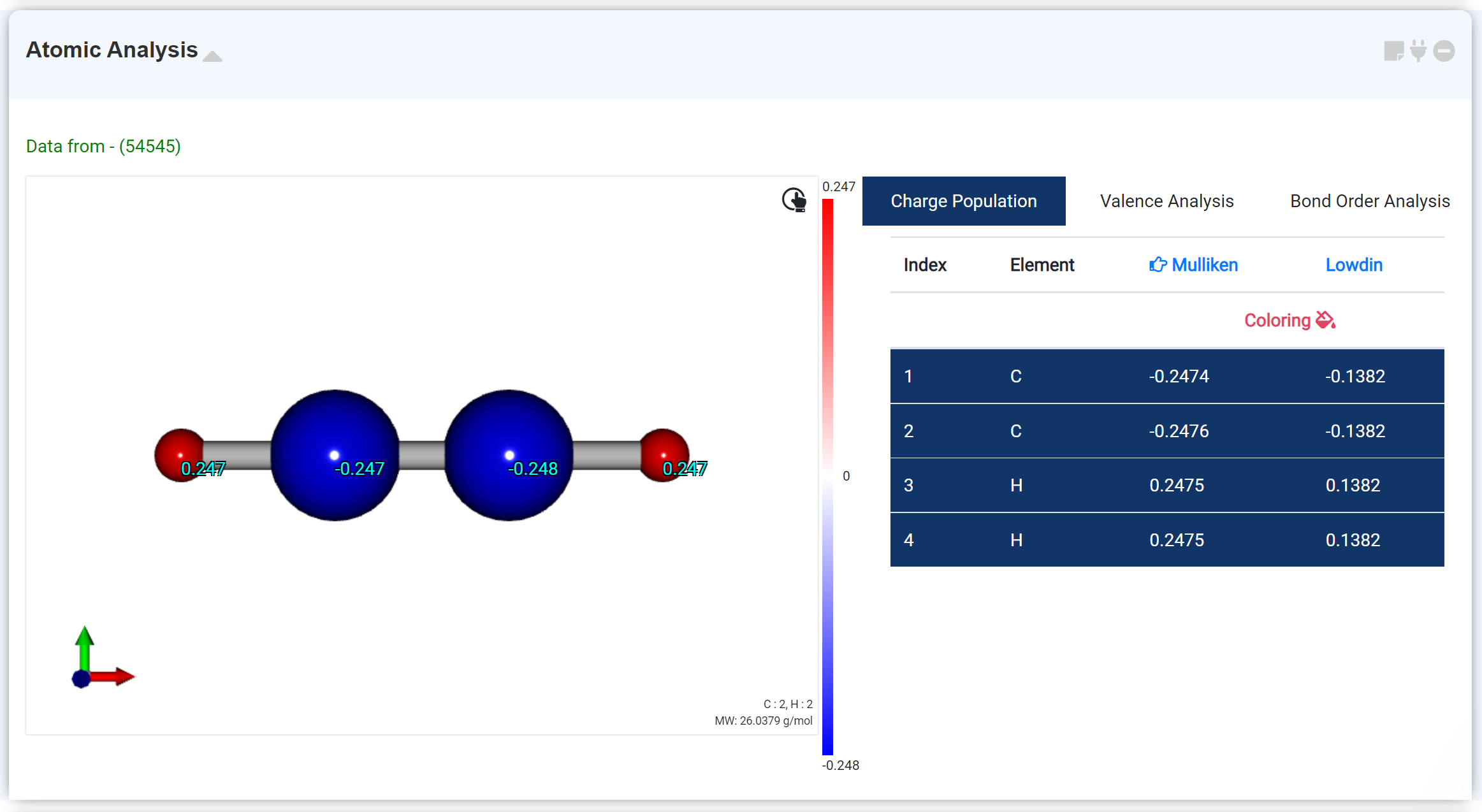
Atomic Analysis 모듈은 분자 좌표 최적화 후 얻어지는 계산결과 중 분자 좌표, 전하, 결합 차수 데이터를 확인하기 위한 모듈입니다.
Charge Population 탭에서는 Mulliken, Lowdin charge를 확인할 수 있습니다. 'Coloring ' 옵션을 클릭하면 전하이동 분포를 색상으로 확인할 수 있습니다. 테이블에서 원하는 원자를 클릭하면 해당 정보가 좌측 분자 구조에 겹쳐 표시됩니다. 헤더 부분의 Mulliken, Lowdin을 클릭하면 원하는 정보를 이용해 coloring하고 charge 데이터를 표시할 수 있습니다.
Valence Analysis 탭에서는 최적화 후 얻어진 Total valence charge, Bonded Valance 데이터를 확인할 수 있습니다.
Bond Order Analysis 탭에서는 각 결합의 결합 차수를 볼 수 있습니다.
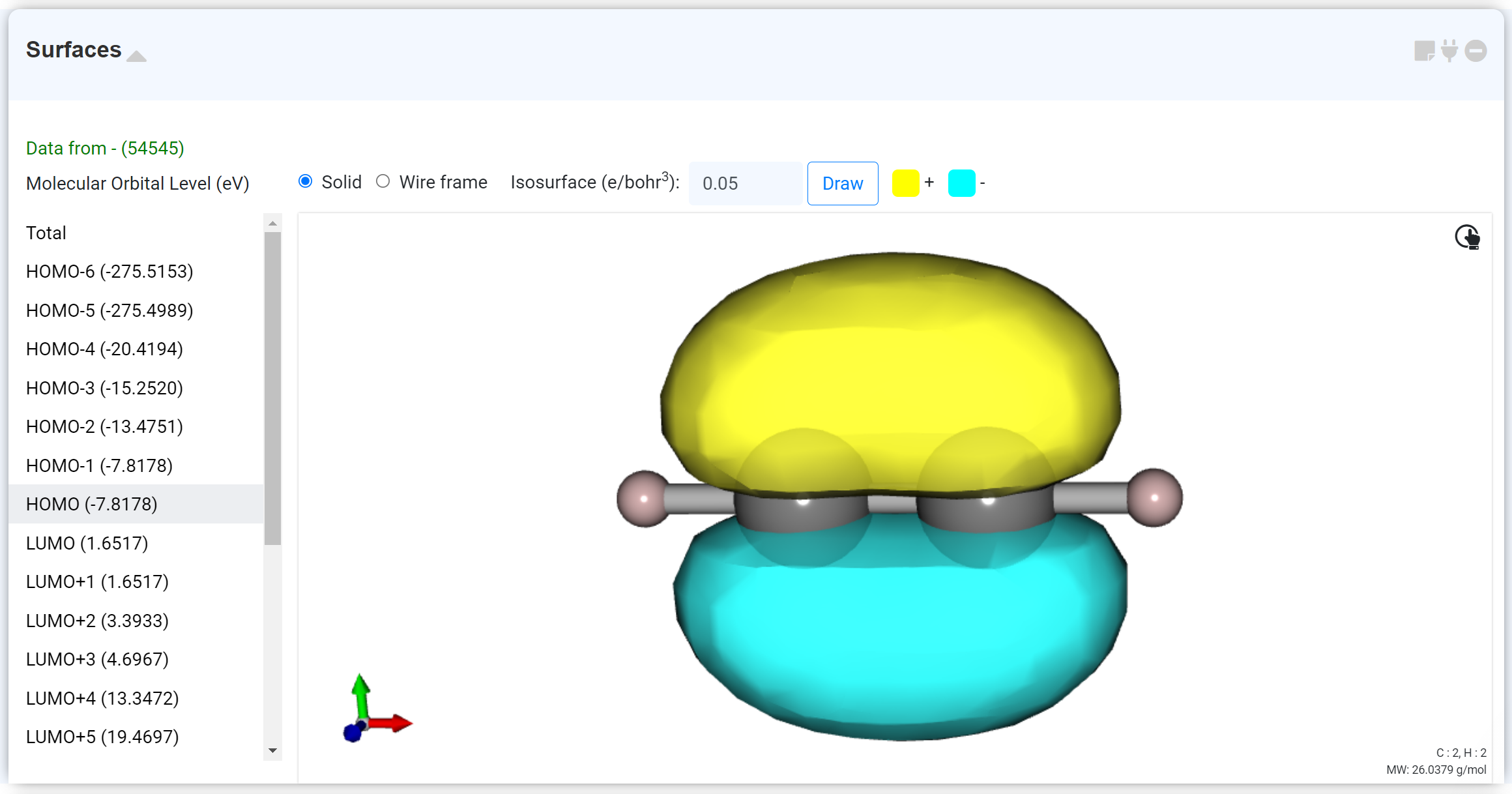
계산된 Molecular orbital을 확인할 수 있습니다. 모듈 추가 시 HOMO가 기본으로 선택되어 있습니다. 상단에서 표시 형식을 Wireframe으로 변경할 수 있으며, Isosurface를 조절할 수 있습니다. Open shell 계산을 수행한 경우, alpha, beta spin을 선택할 수 있습니다.
좌측 리스트에서 MO eigenvalue를 확인할 수 있습니다. 이 데이터는 'Density of States' 모듈에서 그래프 형태로 확인할 수 있습니다.
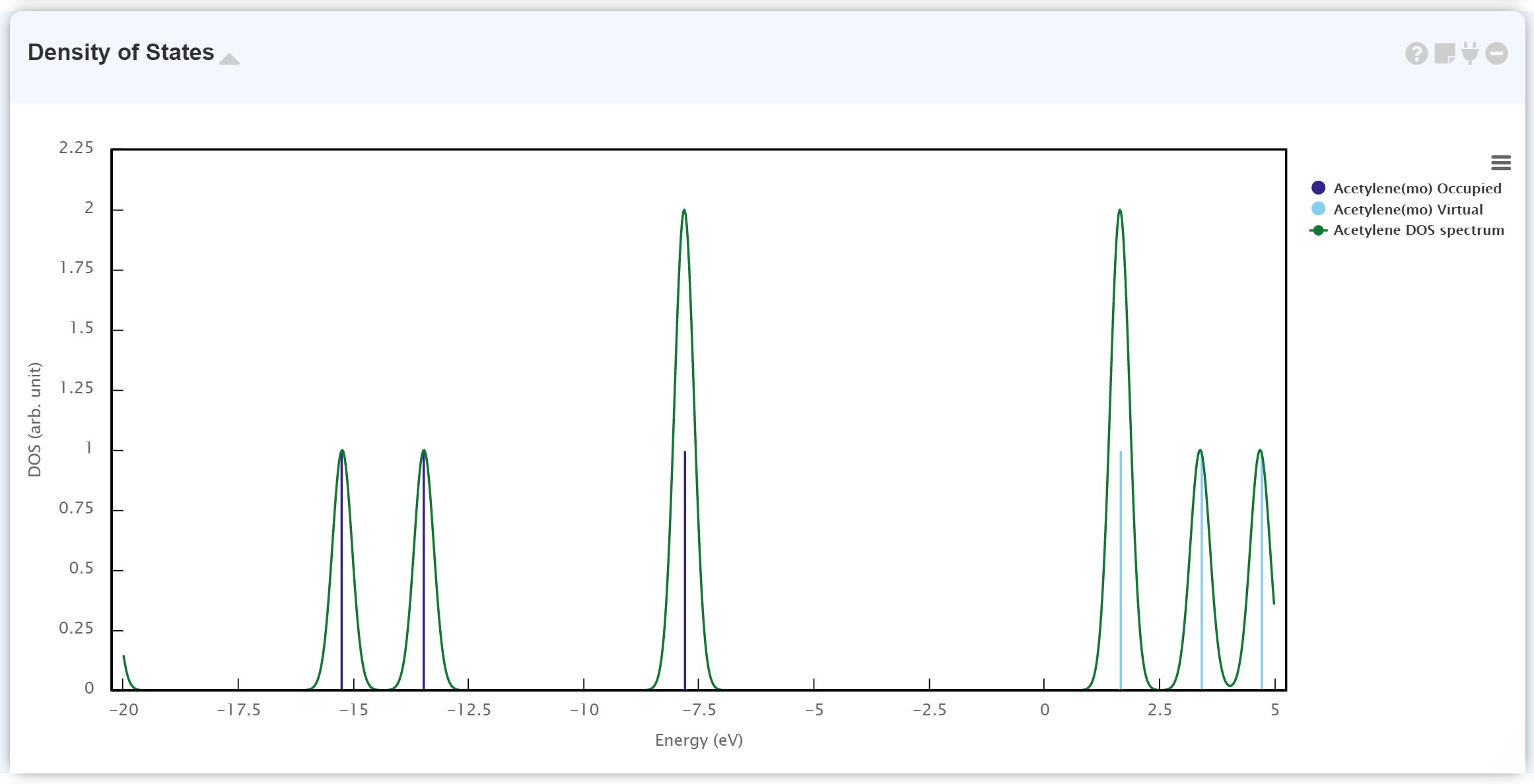
계산된 Electronic Status를 볼 수 있습니다. 표시되는 두 그래프 중 불연속적인 그래프로부터 molecular orbital (MO) eigenvalue를 확인할 수 있습니다. 이때 남색 그래프는 occupied MO이며, 하늘색 그래프는 virtual (unoccupied) MO입니다.
연속적인 선 그래프는 MO eigenvalue에 Gaussian broadening을 가한 데이터입니다.
'Density of States' 모듈과 'Surface' 모듈을 함께 보며 각 MO는 어떠한 모양을 가지고 있는지 확인할 수 있습니다.
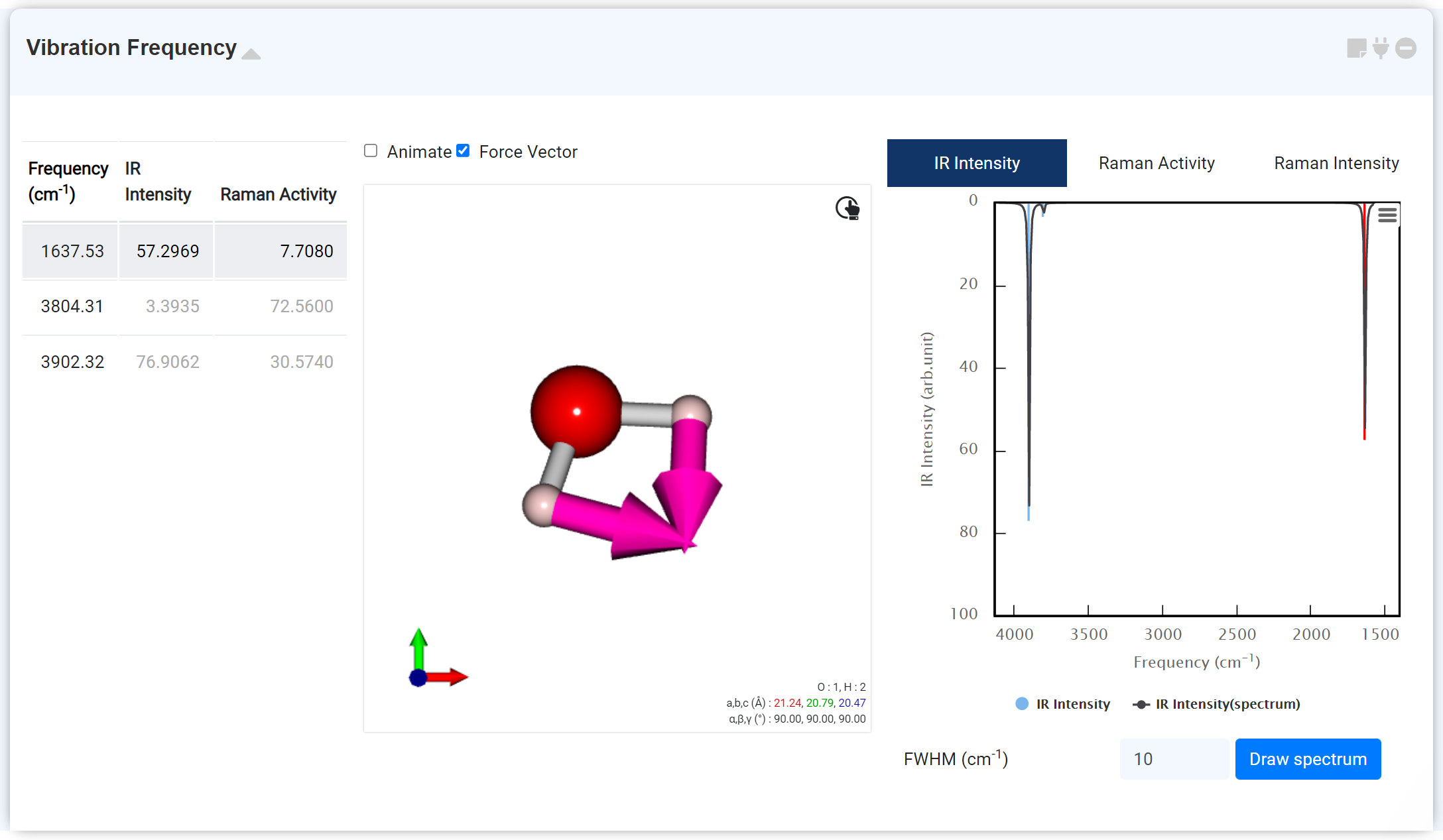
Vibrational Freqency 모듈은 GAMESS 모듈에서 계산한 'IR frequency' 및 ' Raman activity' 데이터를 표시하기 위한 모듈입니다. GAMESS 모듈에서, 'IR frequency'는 'RUN Type: OPTIMIZE' 일 경우 $STATPT 그룹의 'IR frequency' 키워드를 'True'로 설정하여 계산할 수 있습니다. 'Raman activity' 는 Hessian 계산을 수행한 GAMESS 모듈에 새로운 GAMESS 모듈을 연결한 다음 'RUN Type: RAMAN' 계산을 수행하여 얻을 수 있습니다.
Vibrational Frequency 모듈을 추가하면 각 진동 모드 리스트와 해당 진동 모드의 Frequency 정보, 진동 모드 애니메이션, 힘 벡터, 그리고 IR Intensity, Raman Activity, Raman Intensity를 확인할 수 있습니다. 이때, Raman Intensity는 'GaussSum' 을 이용해 Raman activity로부터 변환된 데이터입니다.
필요한 경우, 그래프 하단에서 FWHM (Full Width at Half Maximum, 단위 cm-1)을 변경할 수 있습니다. Raman Intensity의 경우 Excitation Wavelength 및 온도를 변경하여 새로운 스펙트럼을 그릴 수 있습니다.
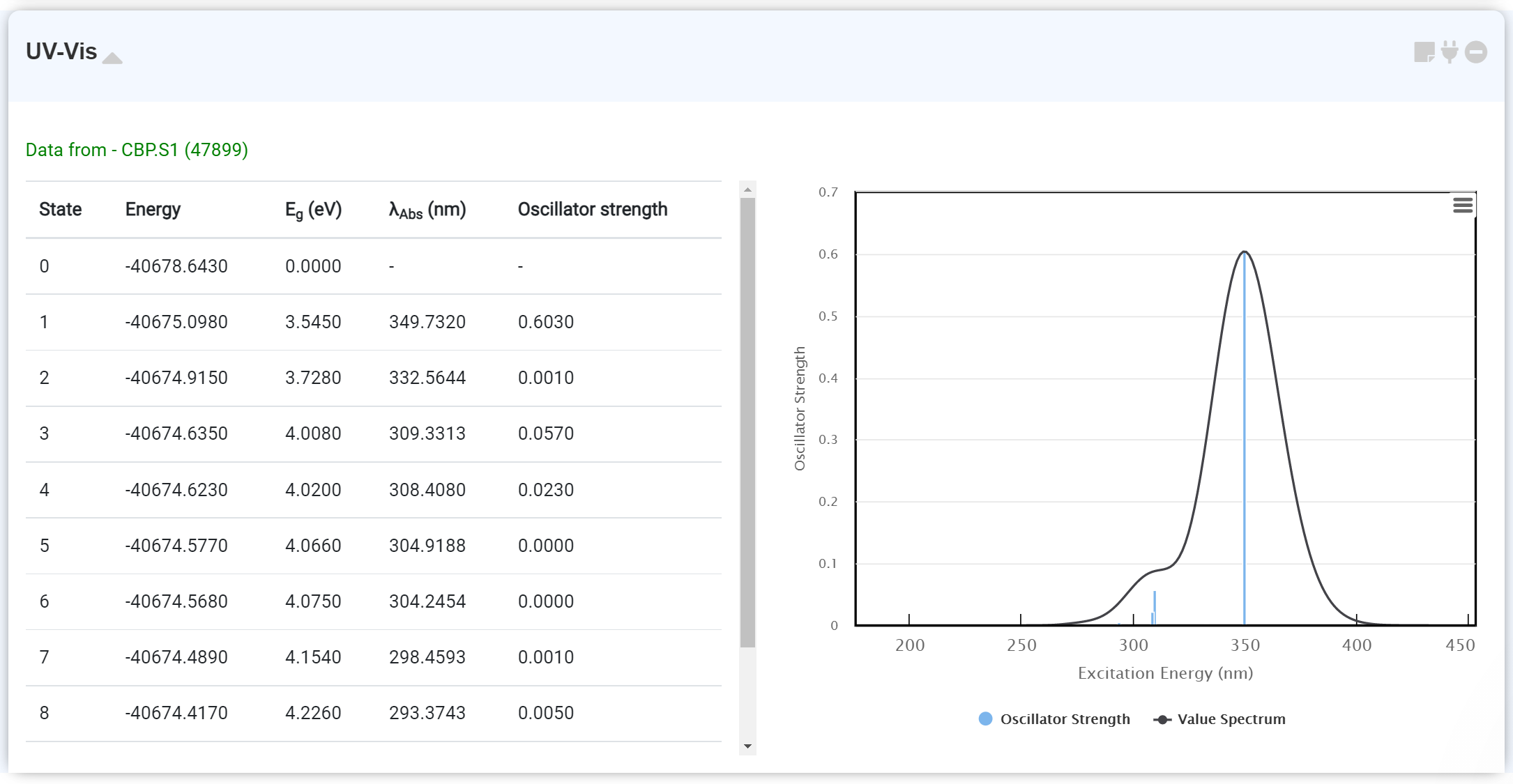
UV/Vis 계산 데이터를 확인할 수 있습니다. GAMESS 모듈에서 'TDDFT: EXCITE' 옵션을 켜고 계산을 수행하면 UV/Vis 데이터를 얻을 수 있습니다. 이때 Optimization을 먼저 수행한 후, 새로운 GAMESS 모듈을 추가하고 기존 optimization 계산을 수행한 GAMESS 모듈에 연결하여 excitation state 계산을 수행하는 것이 좋습니다. 이때 'RUN Type'을 'ENERGY' 로 설정해야 합니다.
우측 스펙트럼에 마우스를 호버하여 λAbs 및 Oscillator Strength를 확인할 수 있습니다.
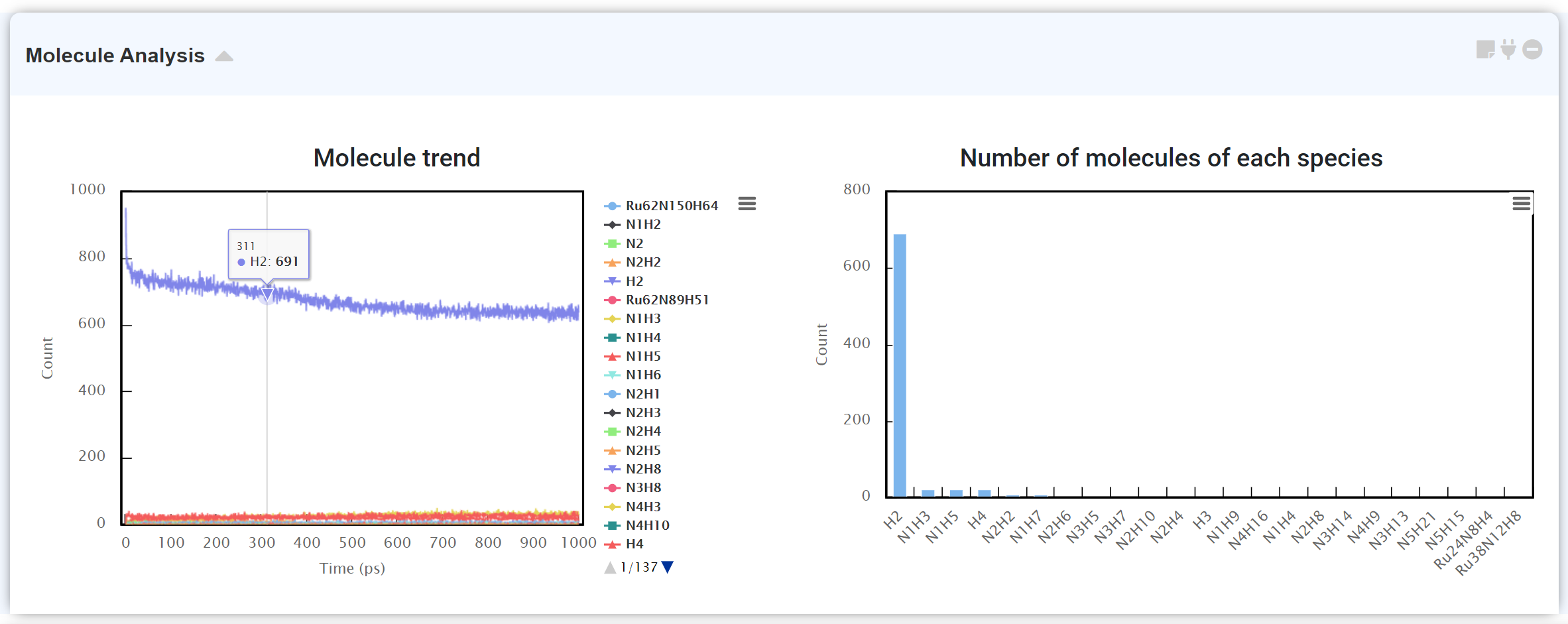
분자동력학(Molecular dynamics) 계산 후, 분자 개수를 세기 위한 후처리 모듈입니다. 화면 왼쪽에는 시스템 내에 존재하는 다양한 분자를 시간에 따라 분석하여 그 개수를 표시합니다. 오른쪽에는 해당 시점에 어떤 분자가 많이 존재하는지 분자별 개수를 보여줍니다.
이때 원자 개수가 많다면 결과 로딩에 시간이 걸릴 수 있습니다.
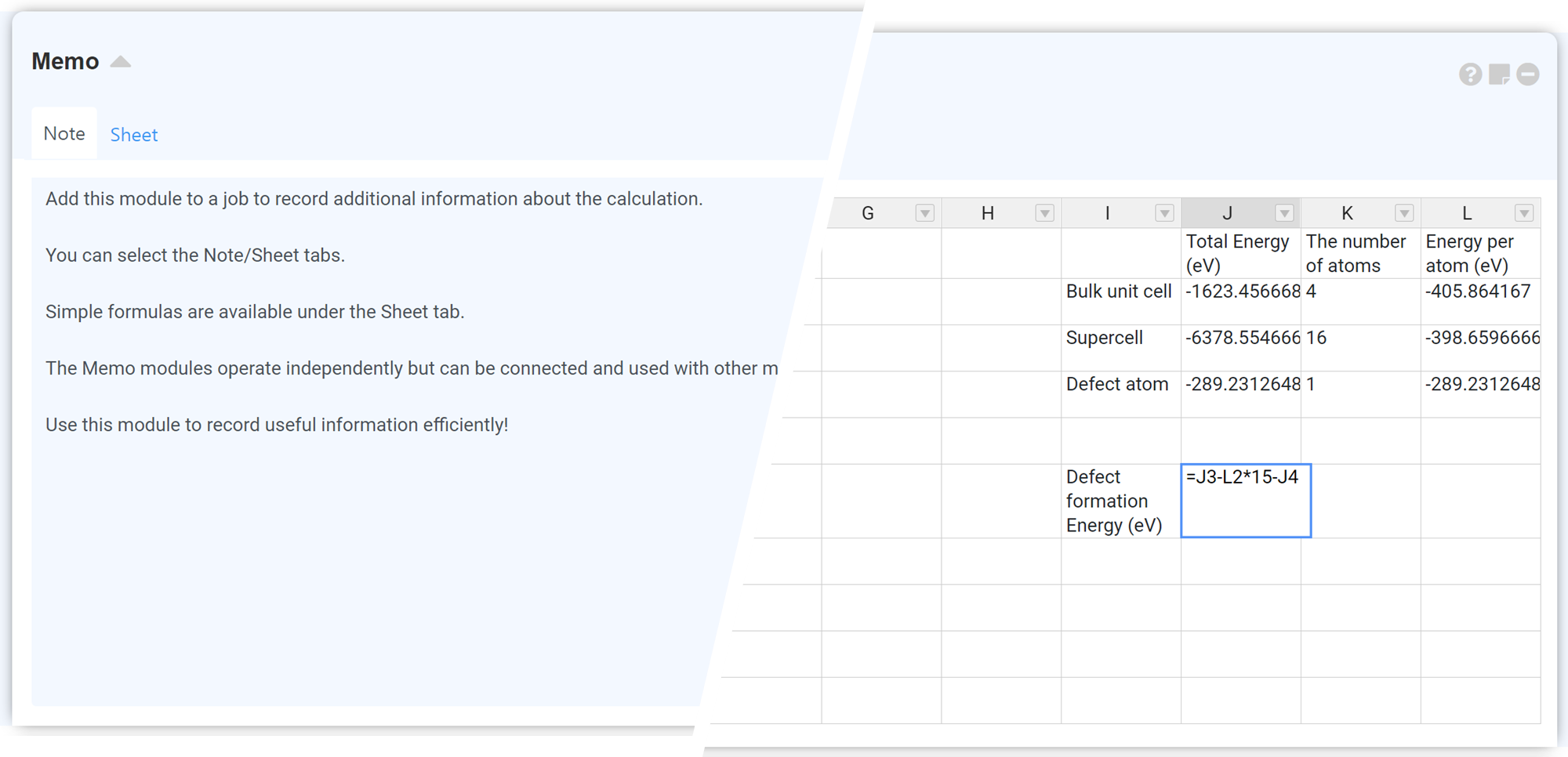
'Sheet' 탭은 Handsontable의 Formulas 플러그인을 사용합니다. 보다 자세한 설명은 Handsontable Formula Support 페이지를 참고하십시오.
기능:
- Any numbers, negative and positive as float or integer;
- Arithmetic operations such as:
+,-,/,*,%,^; - Logical operations such as:
AND(),OR(),NOT(),XOR(); - Comparison operations such as:
=,>,>=,<,<=,<>; - All JavaScript Math constants such as:
PI(),E(),LN10(),LN2(),LOG10E(),LOG2E(),SQRT1_2(),SQRT2(); - Error handling:
#DIV/0!,#ERROR!,#VALUE!,#REF!,#NAME?,#N/A,#NUM!; - String operations such as:
&(concatenation eq.=-(2&5)will return-25); - All excel formulas defined in formula.js;
- Relative and absolute cell references such as:
A1,$A1,A$1,$A$1; - Build-in variables such as:
TRUE,FALSE,NULL; - Custom variables;
- Nested functions;
- Dynamic updates.
알려진 한계점:
- 필터, 열 정렬 기능을 사용할 수 없습니다.
- 행 자르기는 지원되지 않습니다.