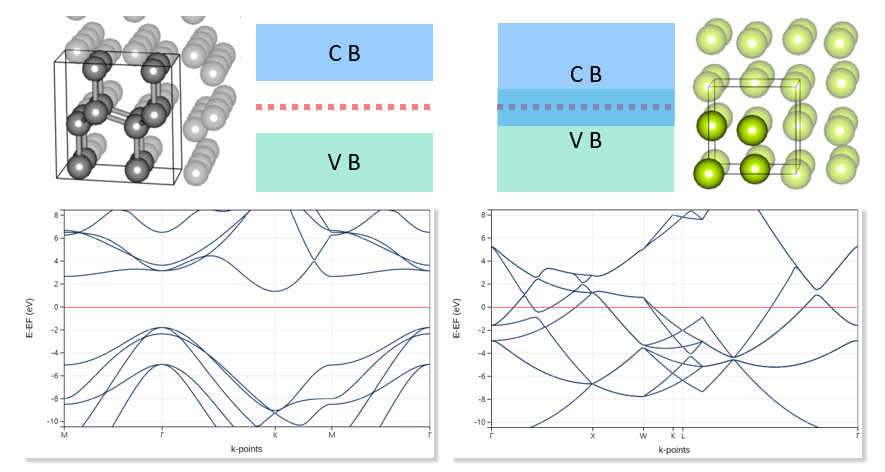
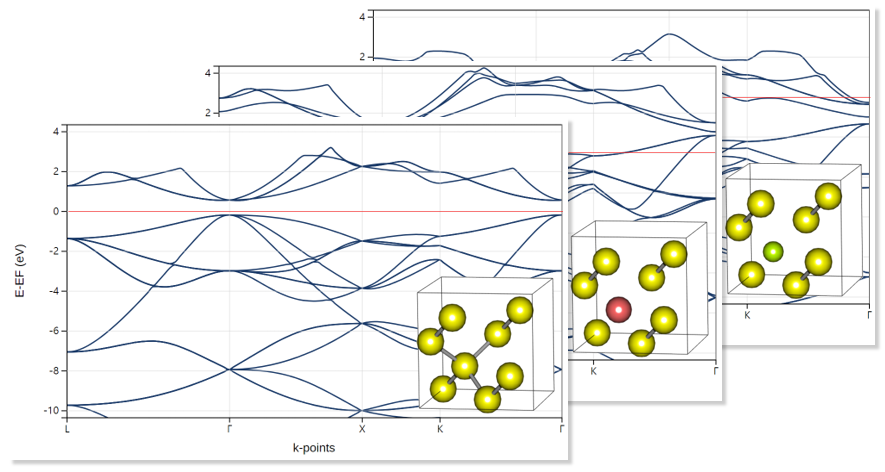
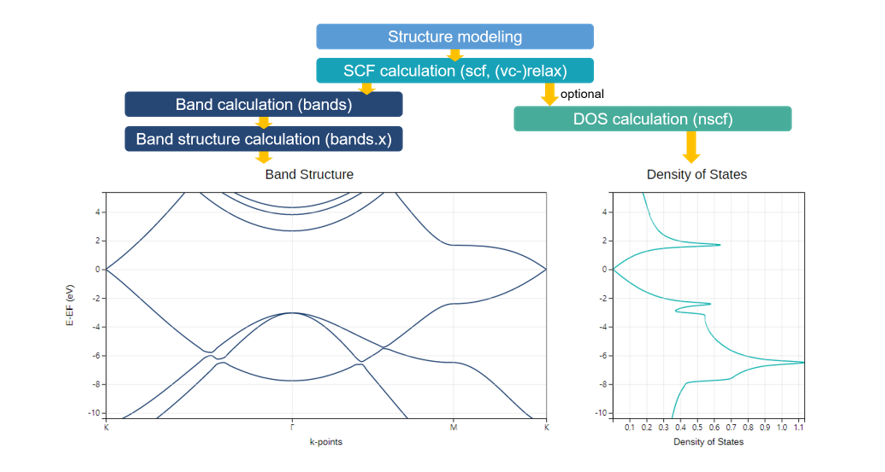
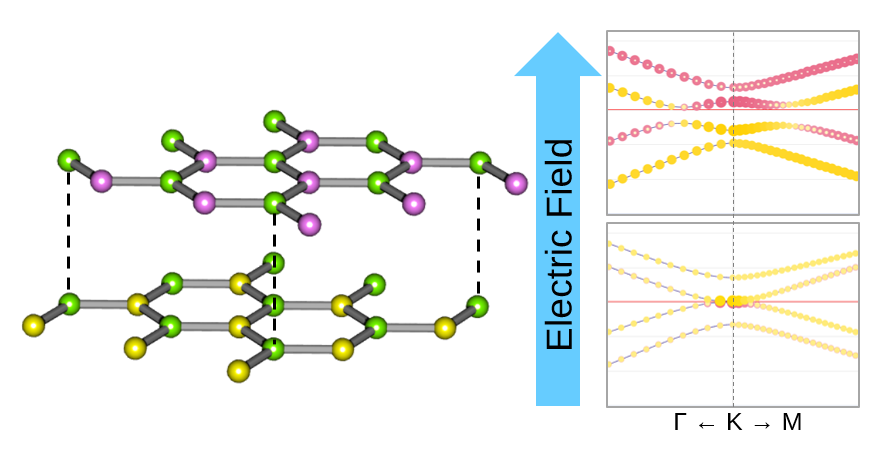
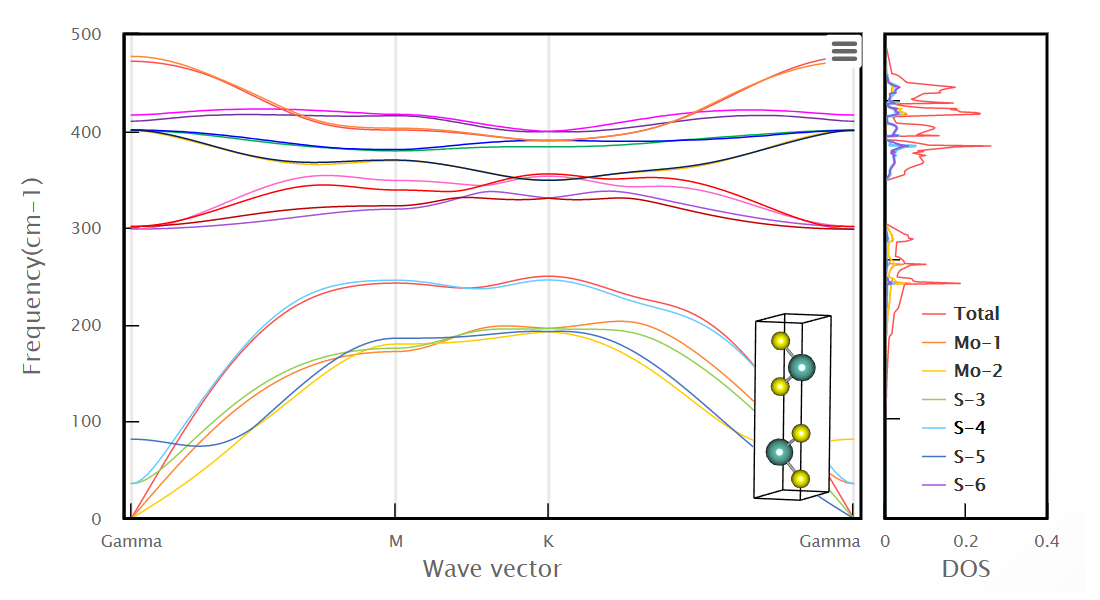
Materials Square Demo
DFT Energy
1. Convergence Test: Cutoff Energy
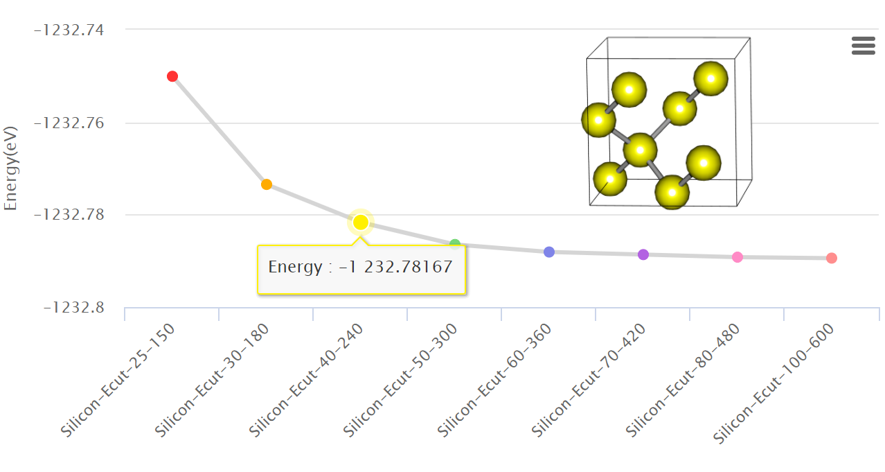
Convergence test is a way of optimizing a simulation to use limited computational resources efficiently. It is important to find a proper initial input setting for simulation research to decide the accuracy and reliability of the simulation. Read more

2. Convergence Test: K-points
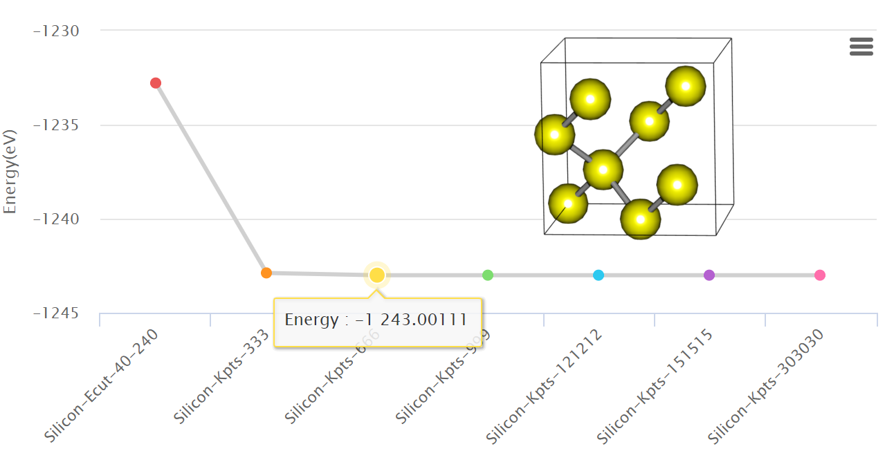
Convergence test is a way of optimizing a simulation to use limited computational resources efficiently. It is important to find a proper initial input setting for simulation research to decide the accuracy and reliability of the simulation. Read more
3. How to Calculate Cohesive Energy
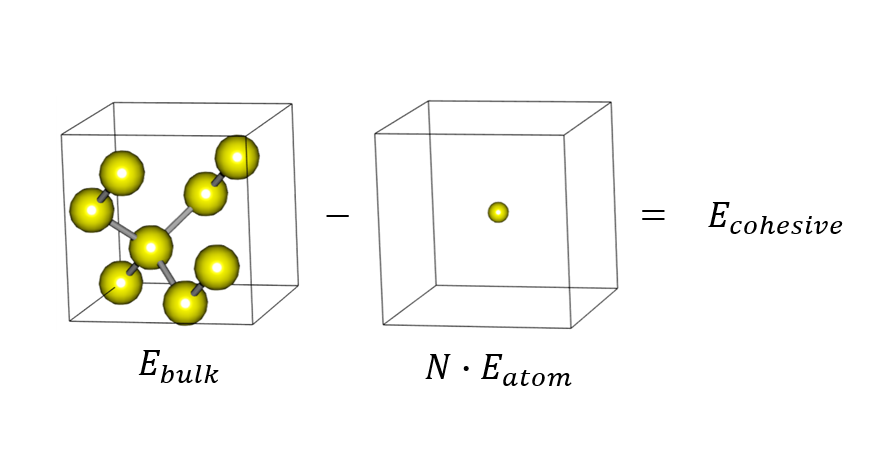
The energy for bonding can be calculated using the difference between a bonded structure and a dissociated one. This energy, which is necessary to separate an atom from the solid, is called cohesive energy. Read more
5. Calculation of a Single Atom
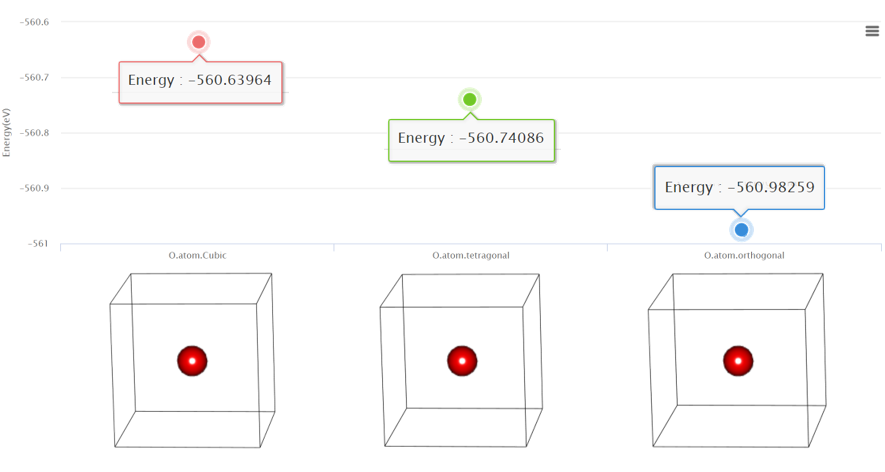
Cohesive energy defined as the difference between the energy per atom in a solid and the energy of a single atom. It can be used to understand the property of crystal such as in finding the correlation effect of an ionic crystal or identifying the stability of the surface. Read more
10-1. Adsorption Energy Obtained through Slab Structure
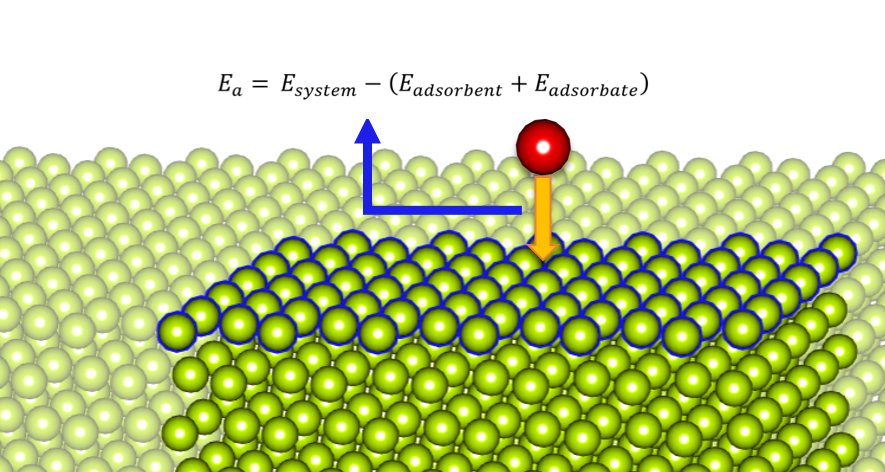
Under the adsorption process, the energy change is called adsorption energy. This energy is mainly used when calculating the chemical engineering properties, exploring the adsorption mechanism, or determining the energetic heterogeneity of the surface of a solid. Read more
10-2. Surface Energy Obtained through Slab Structure
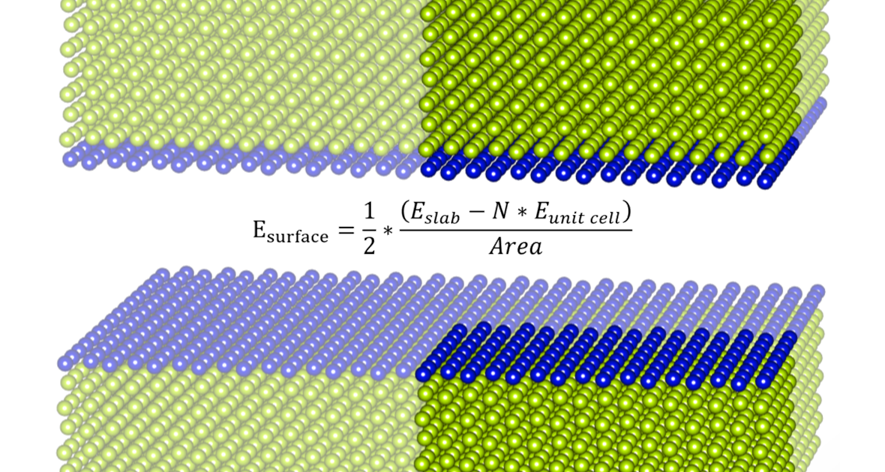
When modeling a slab structure, after the relaxation of the unit cell, use the optimized unit cell to create a slab structure. Then, it can reduce the time for calculating a slab structure. In addition, with the use of the two energies, you can get the surface energy. Read more
11. Stacking Fault Energy
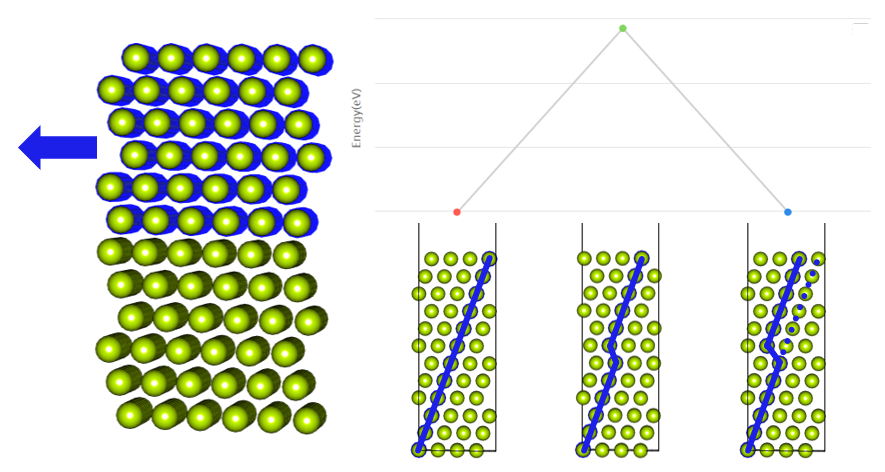
If some of the crystal is moved, a stacking fault is created. The energy generated by the stacking fault is called stacking fault energy (SFE). The stacking fault energy can be used to determine the thermodynamic stability of a crystal structure or to find out the deformation behavior. Read more
16. Bulk modulus
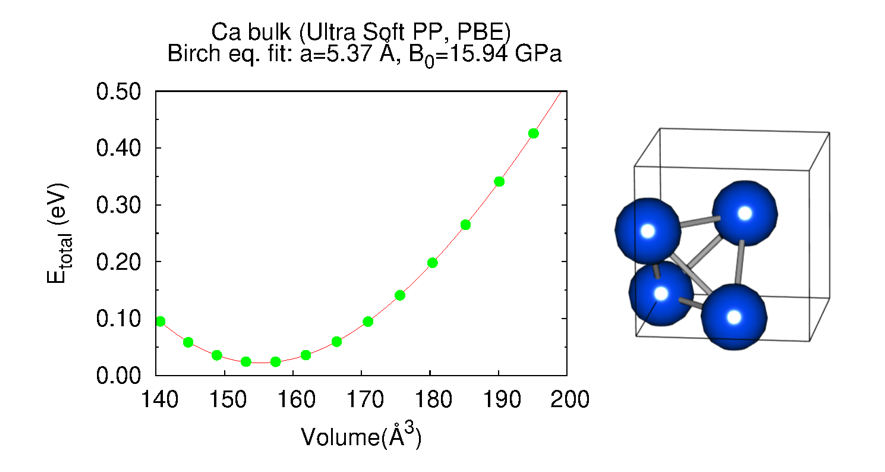
For a material is isotropically compressed, at a small volume change, pressure is proportional to the volume strain, and the proportional constant is called the bulk modulus. The bulk modulus could be calculated by applying the data obtained from the several scf calculations. Read more
19. Cohesive Energy with Different Pseudopotentials
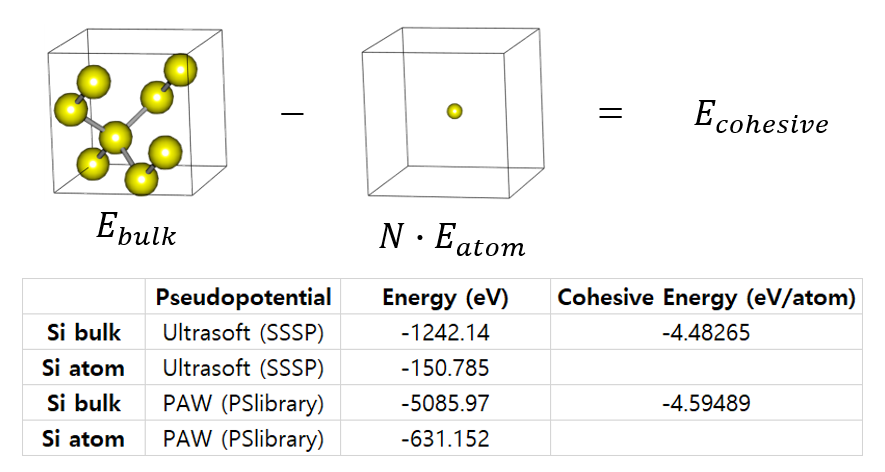
Different pseudopotentials yield different energy values. Therefore, energy values should not be compared directly. But the trend is consistent. So compare the difference in the values. Read more
22. Formation Energy
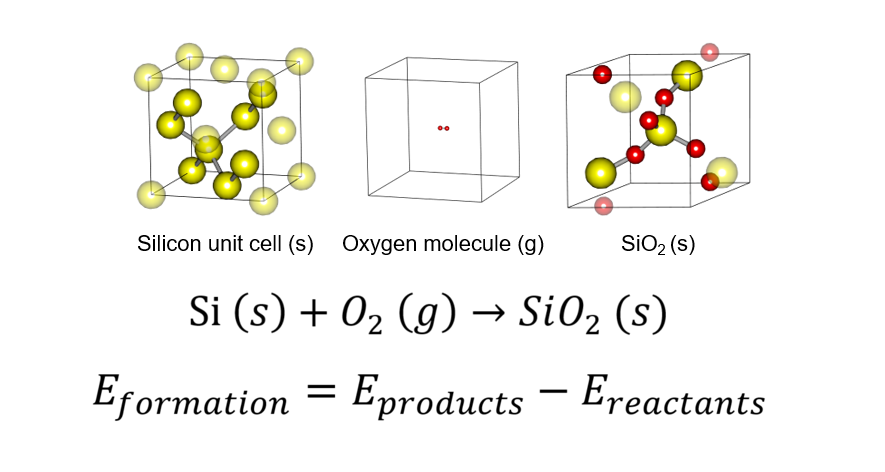
Formation Energy is the value to be used as a measure of the amount of energy entering and exiting a chemical reaction, thus providing a basis for determining thermodynamic stability. Read more
25. Van der Waals Correction
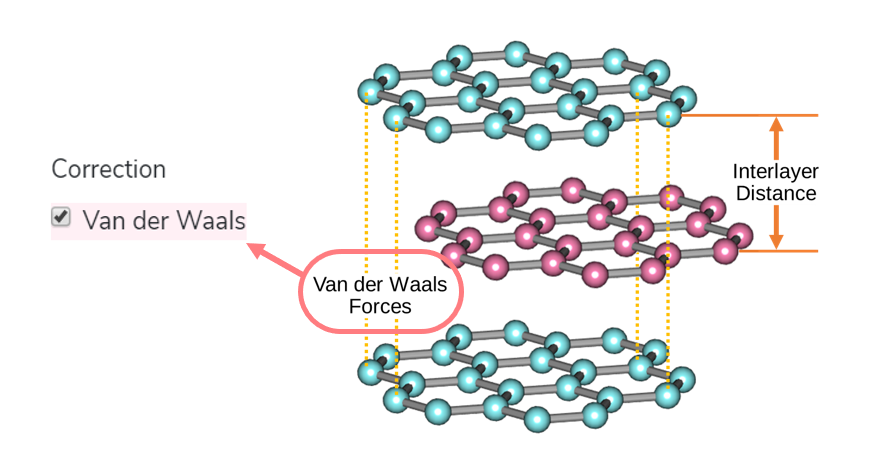
Van der Waals Correction is performed to improve the precision of GGA. Van der Waals Correction, such as DFT+D3, corrects the correlation term of exchange-correlation potential by adding dispersion term. Read more